


 Shyam Diwakar, Ph.D.
Shyam Diwakar, Ph.D.
Assistant Professor, Amrita School of Biotechnology
 Jaap Heringa, Ph.D.
Jaap Heringa, Ph.D.
Director & Professor of Bioinformatics, IBIVU VU University Amsterdam, The Netherlands
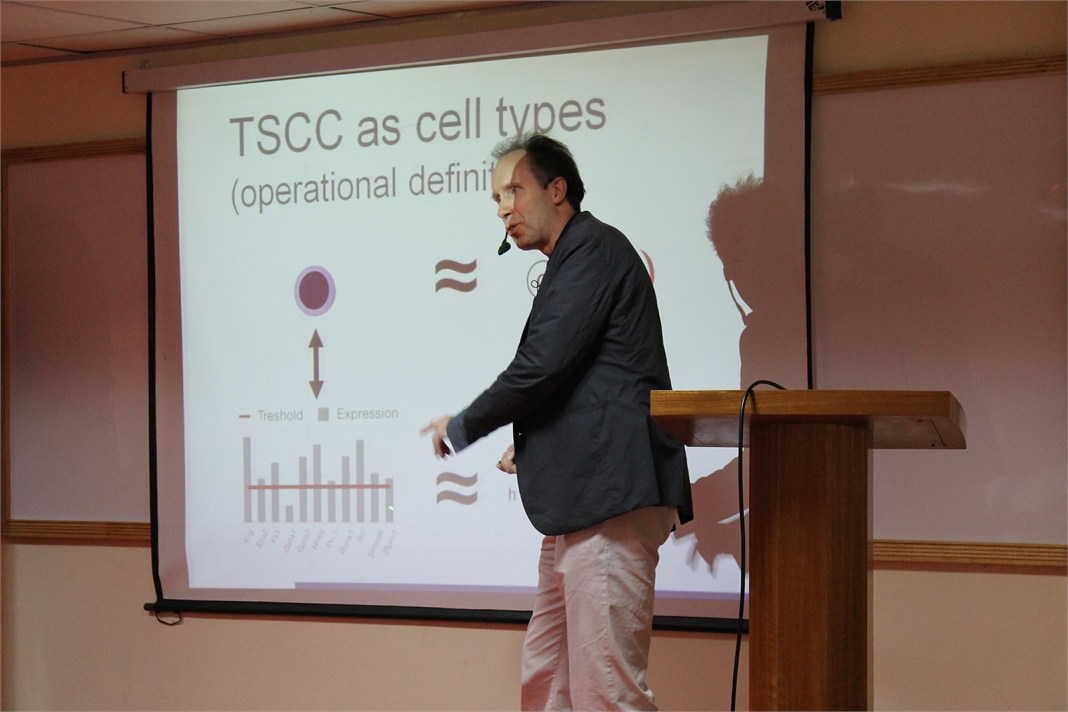
Modeling strategy based on Petri-nets
In my talk I will introduce a formal modeling strategy based on Petri-nets, which are a convenient means of modeling biological processes. I will illustrate the capabilities of Petri-nets as reasoning vehicles using two examples: Haematopoietic stem cell differentiation in mice, and vulval development in C. elegance. The first system was modeled using a Boolean implementation, and the second using a coarse-grained multi-cellular Petri-net model. Concepts such as the model state space, attractor states, and reasoning to adapt the model to the biological reality will be discussed.

 Rajgopal Srinivasan, Ph.D.
Rajgopal Srinivasan, Ph.D.
Principal Scientist & Head Bio IT R&D, TCS Innovation Labs, India
Interpretation of Genomic Variation – Identifying Rare Variations Leading to Disease
Genome sequencing technologies are generating an abundance of data on human genetic variations. A big challenge lies in interpreting the functional relevance of such variations, especially in clinical settings. A first step in understanding the clinical relevance of genetic variations is to annotate the variants for region of occurrence, degree of conservation both within and across species, pattern of variation across related individuals, novelty of the variation and know effects of related variations. Several tools already exist for this purpose. However, these tools have their strengths and weaknesses. A second issue is the development of algorithms, which, given a rich annotation of variants are able to prioritize the variants as being relevant to the phenotype under investigation.
In my talk I will detail work that has been done in our labs to address both of the above problems. I will also illustrate the application of these tools that helped identify a rare mutation in the ATM gene leading to a diagnosis of AT in two infants.
 Ajay Shah, Ph.D.
Ajay Shah, Ph.D. Kal Ramnarayan, Ph.D.
Kal Ramnarayan, Ph.D.
Co-founder President & Chief Scientific Officer, Sapient Discovery, San Diego, CA, USA
A cost-effective approach to Protein Structure-guided Drug Discovery: Aided by Bioinformatics, Chemoinformatics and computational chemistry
With the mapping of the human genome completed almost a decade ago, efforts are still underway to understand the gene products (i.e., proteins) in the human biological and disease pathways. Deciphering such information is very important for the discovery and development of small molecule drugs as well as protein therapeutics for various human diseases for which no cure exists. As an example, with more than 500 members, the kinase family of protein targets continues to be an important and attractive class for drug discovery. While how many of the members in this family are actually druggable is still to be established, there are several ongoing efforts on this class of proteins across a broad spectrum of disease categories. Even though in general the protein structural topology might looks similar, there are issues with respect selectivity of identified small molecule inhibitors when, the lead molecule discovery is carried out at the ATP binding site. As an added complexity, allosteric modulators are needed for some of the members, but the actual site for such modulation on the protein target can not resolved with uncertainty. In this presentation we will describe a bioinformatics and computational based platform for small molecule discovery for protein targets that are involved in protein-protein interactions as well as targets like kinases and phosphatases. We will describe a computational approach in which we have used an informatics based platform with several hundred kinases to sort through in silico and identify inhibitors that are likely to be highly selective in the lead generation phase. We will discuss the implication of this approach on the drug discovery of the kinase and phosphatase classes in general and independent of the disease category.
 Prashanth Athri, Ph.D.
Prashanth Athri, Ph.D.
Senior Specialist, Strand Life Sciences, Bengaluru, India
Rare disease diagnostic platform
At Strand, genomic sequencing combined with bioinformatic analysis have provided discriminative diagnosis in the case of rare genetic disorders. Inspired by these cases, we are building an integrated software that combines curated literature content and bioinformatics databases with a clinically oriented user interface to substantially compress time taken to determine likely candidate genetic variants in a Diagnostic Odyssey. At the back end we employ various algorithms that systematically query our diverse knowledgebase to provide the clinicians a comprehensive, and possibly multidimensional, annotation of the variant in the context of disease.
 Karmeshu, Ph.D.
Karmeshu, Ph.D.
Dean & Professor, School of Computer & Systems Sciences & School of Computational & Integrative Sciences, Jawaharlal Nehru University, India.
Interspike Interval Distribution of Neuronal Model with distributed delay: Emergence of unimodal, bimodal and Power law
The study of interspike interval distribution of spiking neurons is a key issue in the field of computational neuroscience. A wide range of spiking patterns display unimodal, bimodal ISI patterns including power law behavior. A challenging problem is to understand the biophysical mechanism which can generate the empirically observed patterns. A neuronal model with distributed delay (NMDD) is proposed and is formulated as an integro-stochastic differential equation which corresponds to a non-markovian process. The widely studied IF and LIF models become special cases of this model. The NMDD brings out some interesting features when excitatory rates are close to inhibitory rates rendering the drift close to zero. It is interesting that NMDD model with gamma type memory kernel can also account for bimodal ISI pattern. The mean delay of the memory kernels plays a significant role in bringing out the transition from unimodal to bimodal ISI distribution. It is interesting to note that when a collection of neurons group together and fire together, the ISI distribution exhibits power law.
 Lalitha Subramanian, Ph.D.
Lalitha Subramanian, Ph.D.
Chief Scientific Officer & VP, Services at Scienomics, USA
Nanoscale Simulations – Tackling Form and Formulation Challenges in Drug Development and Drug Delivery
Lalitha Subramanian, Dora Spyriouni, Andreas Bick, Sabine Schweizer, and Xenophon Krokidis Scienomics
The discovery of a compound which is potent in activity against a target is a major milestone in Pharmaceutical and Biotech industry. However, a potent compound is only effective as a therapeutic agent when it can be administered such that the optimal quantity is transported to the site of action at an optimal rate. The active pharmaceutical ingredient (API) has to be tested for its physicochemical properties before the appropriate dosage form and formulation can be designed. Some of the commonly evaluated parameters are crystal forms and polymorphs, solubility, dissolution behavior, stability, partition coefficient, water sorption behavior, surface properties, particle size and shape, etc. Pharmaceutical development teams face the challenge of quickly and efficiently determining a number of properties with small quantities of the expensive candidate compounds. Recently the trend has been to screen these properties as early as possible and often the candidate compounds are not available in sufficient quantities. Increasingly, these teams are leveraging nanoscale simulations similar to those employed by drug discovery teams for several decades. Nanoscale simulations are used to predict the behavior using very little experimental data and only if this is promising further experiments are done. Another aspect where nanoscale simulations are being used in drug development and drug delivery is to get insights into the behavior of the system so that process failures can be remediated and formulation performance can be improved. Thus, the predictive screening and the in-depth understanding leads to experimental efficiency resulting in far-reaching business impacts.
With specific examples, this talk will focus on the different types of nanoscale simulations used to predict properties of the API in excipients and also provide insight into system behavior as a function of shelf life, temperature, mechanical stress, etc.
 Srisairam Achuthan, Ph.D.
Srisairam Achuthan, Ph.D.
Senior Scientific Programmer, Research Informatics Division, Department of Information Sciences, City of Hope, CA, USA
Applying Machine learning for Automated Identification of Patient Cohorts
Srisairam Achuthan, Mike Chang, Ajay Shah, Joyce Niland
Patient cohorts for a clinical study are typically identified based on specific selection criteria. In most cases considerable time and effort are spent in finding the most relevant criteria that could potentially lead to a successful study. For complex diseases, this process can be more difficult and error prone since relevant features may not be easily identifiable. Additionally, the information captured in clinical notes is in non-coded text format. Our goal is to discover patterns within the coded and non-coded fields and thereby reveal complex relationships between clinical characteristics across different patients that would be difficult to accomplish manually. Towards this, we have applied machine learning techniques such as artificial neural networks and decision trees to determine patients sharing similar characteristics from available medical records. For this proof of concept study, we used coded and non-coded (i.e., clinical notes) patient data from a clinical database. Coded clinical information such as diagnoses, labs, medications and demographics recorded within the database were pooled together with non-coded information from clinical notes including, smoking status, life style (active / inactive) status derived from clinical notes. The non-coded textual information was identified and interpreted using a Natural Language Processing (NLP) tool I2E from Linguamatics.

Kunal Kundu, Sushma Motamarri, Uma Sunderam, Steven E. Brenner and Rajgopal Srinivasan.
VARANT: The Variant Annotation Tool
Genome sequencing technologies are generating an abundance of data on human genetic variations. A big challenge lies in interpreting the functional relevance of such variations, especially in clinical settings. A first step in understanding the clinical relevance of genetic variations is to annotate the variants for region of occurrence, degree of conservation both within and across species, pattern of variation across related individuals, novelty of the variation and know effects of related variations. Several tools already exist for this purpose. However, these tools have their strengths and weaknesses. We will present an open-source tool, VARANT, written in the python programming language, that is easily extended to incorporate newer annotations.
A detailed variant annotation places variants in context, highlights significant findings and prioritizes candidates for further analysis. With this outlook we developed VARANT to annotate, prioritize and visualize variants. VARANT has 5 levels of annotation – genomic position based, gene based, untranslated region (UTR) based, mutation effect prediction and gene level disease association. The databases used for annotations have been compiled from several sources. The genomic position based annotation comprises of tagging variants present in dbSNP and 1000 Genomes projects, GWAS variants, variants in functionally constrained region and variants overlapping epigenetic signals. The gene-based annotation includes, the distance from splice sites for intronic variants; gene, transcript, amino acid change and splicing silencer and enhancers information for exonic variants. UTR based annotations comprise of UTR functional sites like miRNA binding site, internal ribosomal entry site, variations and deletions in UTR5-Coding Sequence(CDS) boundary, exon-intron boundary and CDS-UTR3 boundary.Mutation effect predictions are incorporated from PolyPhen2 and SIFT. Thus, a detailed annotation with VARANT captures multiple biological aspects of a variant and helps in filtering variants based on disease context. The input and output of VARANT is the universal Variant Call Format, with facilities to export the annotations to popular formats such as comma/tab separated values and MS Excel. Using a desktop computer with single core and 4GB RAM VARANT annotates over 50,000 variants/minute and can be readily parallelized. Being an exhaustive annotator with good performance using modest computational hardware, VARANT is a useful annotation tool for analyzing genomic variants. Furthermore, the tool includes facilities to update the underlying data sources in an automated fashion, and is easily extended to add additional annotations. VARANT also provides an interface to visualize variants in an annotated VCF file and to filter variants interactively based on annotation features like – region, mutation effect etc, and inheritance models. In addition to annotation, there are ongoing efforts to incorporate a variant prioritization module using the annotated features as well as inheritance information.
Bhadrachalam Chitturi, Balaji Raghavachari and Donghyun Kim
Efficient gene prioritization
The gene prioritization, GP, problem seeks to identify the most promising genes among several candidate genes. In genetics, gene related conditions are typically associated with chromosomal regions, say with GWAS. These associations yield lists of candidate genes. A priori, some genes i.e. seed genes, are associated with a specific disease D; additional genes that are implicated via associations constitute the potential candidates. Thus, most promising novel candidates for D are sought. In network based approach, a protein protein interaction network, i.e. NP , and a set S of seed genes constitute the prior knowledge. We treat a gene and the protein that it encodes identically. Various GP algorithms based on guilt by association are run on the NP to predict novel candidates [1–6]. They rank a new candidate gene by its estimated association to D.
Distance between a pair of genes is the shortest path measured in the number of edges. Diameter of a set of genes is the longest distance between any pair of genes in terms of the number of edges. The density of a set X of genes is defined as e(X)/|X| where e(X) denotes the number of edges among genes of X and |X| denotes the number of genes of X. The set S: (i) can be of minimal size (say one), (ii) is tightly coupled in NP , i.e. has low-diameter/high-density, or (iii) is loosely coupled, i.e. has high-diameter/low-density. Similarly, the GP algorithms can be partitioned into: Type-1 that ignore the edge weights and Type-2 that employ the edge weights. However, currently, the prioritization process neither exploits the character of S nor the type of GP algorithm that is run. Given S, we compute two core networks of NP which we call NC1 and NC2 that are subnetworks of NP . The idea is to execute GP algorithms of Type-1 and Type-2 on NC1 and NC2 respectively instead of NP . Typically, NC1 and NC2 are much smaller than NP . Also, one runs several algorithms of Type-1 and Type-2 [2–4, 6] and takes consensus [6].
In general, the time to run a GP algorithm say AP on NP i.e. t1 or to compute NC1 and NC2 i.e. t2 is proportional to e(NP ) where e(NP ) e(NC1) and e(NP ) e(NC2). However, executing AP on NC1/NC2 (a much smaller network) is much more efficient than executing AP on NP . We run several GP algorithms onNC1/NC2 [6] but computeNC1/NC2 only once. So, overall our method is more efficient. Preliminary implementation results show that for several GP algorithms, the candidates identified by our method match the topmost prioritized candidates identified by the direct execution of the algorithm on NP . Overall, our method was more efficient. Based on the number of candidates that we seek and the nature of S, we can generate variants of NCx, x ∈ {1, 2}. In some cases, AP determines the appropriate variant of NCx.
Nitish Sathyanrayanan, Sandesh Ganji and Holenarsipur Gundurao Nagendra.
Insilico Analysis of hypothetical proteins from Leishmania donovani: A Case study of a membrane protein of the MFS class reveals their plausible roles in drug resistance
Kala-azar or visceral leishmaniais (VL), caused by protozoan parasite Leishmania donovani, is one of the leading causes of morbidity and mortality in Bihar, India (Guerin et al. 2002; Mubayi et al. 2010). The disease is transmitted to the humans mainly by the vector, Phlebotmus argentipes, commonly known as Sand fly. The majority of VL (> 90%) occurs in only six countries: Bangladesh, India, Nepal, Sudan, Ethiopia and Brazil (Chappuis et al. 2007). In the Indian subcontinent, about 200 million people are estimated to be at risk of developing VL and this region harbors an estimated 67% of the global VL disease burden. The Bihar state only has captured almost 50% cases out of total cases in Indian sub-continent (Bhunia et al. 2013). ‘Conserved hypothetical’ proteins pose a challenge not just to functional genomics, but also to biology in general (Galperin and Koonin 2004). Leishmania donovani (strain BPK282A1) genome consists of a staggering ∼65% of hypothetical proteins. These uncharacterized proteins may enable better appreciation of signalling pathways, general metabolism, stress response and even drug resistance.
Rajasekhar Chekkara, Venkata Reddy Gorla and Sobha Rani Tenkayala
Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors
Polo-like kinase 1 (PLK1) is a significant enzyme with diverse biological actions in cell cycle progression, specifically mitosis. Suppression of PLK1 activity by small molecule inhibitors has been shown to inhibit cancer, being BI 2536 one of the most potent active inhibitor of PLK1 mechanism. Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies were carried out for a set of 54 compounds belonging to Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK1 inhibitors. A six-point pharmacophoremodel AAADDR, with three hydrogen bond acceptors (A), two hydrogen bond donors (D) and one aromatic ring (R) was developed by Phase module of Schrdinger suite Maestro 9. The generated pharmacophore model was used to derive a predictive atom-based 3D quantitative structure-activity relationship analysis (3D-QSAR) model for the training set (r2 = 0.88, SD = 0.21, F = 57.7, N = 44) and for test set (Q2 = 0.51, RMSE = 0.41, PearsonR = 0.79, N = 10). The original set of compounds were docked into the binding site of PLK1 using Glide and the active residues of the binding site were analyzed. The most active compound H18 interacted with active residues Leu 59, Cys133 (glide score = −10.07) and in comparison of BI 2536, which interacted with active residues Leu 59, Cys133 (glide score = −10.02). The 3D-QSAR model suggests that hydrophobic and electron-withdrawing groups are essential for PLK1 inhibitory activity. The docking results describes the hydrogen bond interactions with active residues of these compounds. These results which may support in the design and development of novel PLK1 inhibitors.


















Dr. Bipin Nair,
Dean-Biotechnology, Amrita University
 Vural Özdemir, MD, Ph.D., DABCP
Vural Özdemir, MD, Ph.D., DABCP
Co-Founder, DELSA Global, Seattle, WA, USA
Crowd-Funded Micro-Grants to Link Biotechnology and “Big Data” R&D to Life Sciences Innovation in India
Vural Özdemir, MD, PhD, DABCP1,2*
- Data-Enabled Life Sciences Alliance International (DELSA Global), Seattle, WA 98101, USA;
- Faculty of Management and Medicine, McGill University, Canada;
ABSTRACT
Aims: This presentation proposes two innovative funding solutions for linking biotechnology and “Big Data” R&D in India with artisan small scale discovery science, and ultimately, with knowledge-based innovation:
- crowd-funded micro-grants, and
- citizen philanthropy
These two concepts are new, and inter-related, and can be game changing to achieve the vision of biotechnology innovation in India, and help bridge local innovation with global science.
Background and Context: Biomedical science in the 21(st) century is embedded in, and draws from, a digital commons and “Big Data” created by high-throughput Omics technologies such as genomics. Classic Edisonian metaphors of science and scientists (i.e., “the lone genius” or other narrow definitions of expertise) are ill equipped to harness the vast promises of the 21(st) century digital commons. Moreover, in medicine and life sciences, experts often under-appreciate the important contributions made by citizen scholars and lead users of innovations to design innovative products and co-create new knowledge. We believe there are a large number of users waiting to be mobilized so as to engage with Big Data as citizen scientists-only if some funding were available. Yet many of these scholars may not meet the meta-criteria used to judge expertise, such as a track record in obtaining large research grants or a traditional academic curriculum vitae. This presentation will describe a novel idea and action framework: micro-grants, each worth $1000, for genomics and Big Data. Though a relatively small amount at first glance, this far exceeds the annual income of the “bottom one billion” – the 1.4 billion people living below the extreme poverty level defined by the World Bank ($1.25/day).
We will present two types of micro-grants. Type 1 micro-grants can be awarded through established funding agencies and philanthropies that create micro-granting programs to fund a broad and highly diverse array of small artisan labs and citizen scholars to connect genomics and Big Data with new models of discovery such as open user innovation. Type 2 micro-grants can be funded by existing or new science observatories and citizen think tanks through crowd-funding mechanisms described herein. Type 2 micro-grants would also facilitate global health diplomacy by co-creating crowd-funded micro-granting programs across nation-states in regions facing political and financial instability, while sharing similar disease burdens, therapeutics, and diagnostic needs. We report the creation of ten Type 2 micro-grants for citizen science and artisan labs to be administered by the nonprofit Data-Enabled Life Sciences Alliance International (DELSA Global, Seattle: http://www.delsaglobal.org). Our hope is that these micro-grants will spur novel forms of disruptive innovation and life sciences translation by artisan scientists and citizen scholars alike.
Address Correspondence to:
Vural Özdemir, MD, PhD, DABCP
Senior Scholar and Associate Professor
Faculty of Management and Medicine, McGill University
1001 Sherbrooke Street West
Montreal, Canada H3A 1G5
Email: vural.ozdemir@alumni.utoronto.ca




The global healthcare scene of which the pharmaceutical industry and its products are integral components is today at the cross roads. The high and unaffordable costs of drug research with estimates of over 1 billion dollars for every new drug discovered and developed, the very low success rates, the high degree of obsolescence due to undesirable adverse drug reactions, the decline in the development pipeline of new drugs, patent expiries leading to generic competition and the public’s disillusionment with use of chemicals for human consumption as drugs have all significantly contributed to the problems of this lifeline industry. The strategy adopted by the large R&D based Corporations to get bigger and bigger through mergers and acquisitions to improve cost-effectiveness and productivity of R&D has so far not worked effectively. Consequently, one of the recent trends in healthcare, articulated by many experts is to look for alternate or even complementary approaches to reduce the impact of rising costs of drugs on healthcare. Various new strategies for drug discovery such as the use of Natural Products especially medicinal plants are being actively pursued by healthcare planners and providers. Side by side, traditional systems of medicine whether from the oriental countries or the western nations are also having a serious relook to understand their usefulness in healthcare. To achieve its legitimate position in the healthcare scenario, it is essential to scientifically validate their claimed utility through appropriate and systematic research efforts including pre-clinical and clinical studies. In addition to their own use as medicines, knowledge on the Indian Traditional Medicines can be used as a platform for new drug discovery. The huge potential for carrying out systematic R&D programs for new Drug Discovery based on natural products and possible strategies to realise them in the coming decades will be explained in this presentation.

 Ramani A. Aiyer, Ph.D., MBA
Ramani A. Aiyer, Ph.D., MBA
Principal, Shasta BioVentures, San Jose, CA, USA
New Drug R&D in India: Challenges & Opportunities
New drug discovery and development has become a global endeavor, with Western big pharmaceutical companies farming out more and more chemistry and biology research to Asia, particularly India and China. During the last decade, several Indian pharmaceutical companies have embarked on ambitious R&D programs, with slow but steady progress in developing new chemical / molecular entities. The Indian government has also made a strong commitment to promote innovation and entrepreneurship in the biotechnology sector. The first part of the talk will focus on a case study showing the entire process of discovery and development of a new drug recently launched for Rheumatoid Arthritis. We will then address the challenges of conducting innovative R&D in India and actions necessary to overcome them. The second part of the talk will make the case for developing Ayurvedic drug formulations for the Western / Global markets, again using the example of Rheumatoid Arthritis (Aamavaata). Ayurveda takes a holistic approach to disease diagnosis and therapy based on interactions among body type (prakriti), tri-doshas (three body humors), sapta-dhatus (seven tissues) and malas (excretions). The drugs prescribed are usually herbo-mineral formulations comprising multiple medicinal plants and / or metals. The manufacturing processes date back to Ayurvedic texts several thousand years old, and are compiled in the Ayurvedic Pharmacopeia. Also, the treatment modalities and drug formulations are “personalized” to fit different patient types, based on the holistic diagnoses mentioned earlier. There is a tremendous need to establish a sound basis for Ayurvedic drug discovery R&D for the modern world. We must find a scientific and ethical way to leverage the vast body of anecdotal and possibly retrospective data on patients undergoing Ayurvedic treatment. Combined with in vitro and in vivo biological data on Ayurvedic herbo-mineral formulations, the adoption of stringent manufacturing practices, and designing sound clinical trials to establish the safety and efficacy, India has a golden opportunity to expand the reach of Ayurvedic drugs into Western / Global medical practice.

Sukhithasri V, Nisha N, Vivek V and Raja Biswas
The host innate immune system acts as the first line of defense against invading pathogens. During an infection, the host innate immune cells recognize unique conserved molecules on the pathogen known as Pathogen Associated Molecular Patterns (PAMPs). This recognition of PAMPs helps the host mount an innate immune response leading to the production of cytokines (Akira et al. 2006). Peptidoglycan, one of the most conserved and essential component of the bacterial cell wall is one such PAMP. Peptidoglycan is known to have potent proinflammatory properties (Gust et al. 2007). Host recognize peptidoglycan using Nucleotide oligomerization domain proteins (NODs). This recognition of peptidoglycan activates the NODs and triggers downstream signaling leading to the nuclear translocation of NF-κB and production of cytokines (McDonald et al. 2005). Pathogenic bacteria modify their peptidoglycan as a strategy to evade innate immune recognition, which helps it to establish infection in the host. These peptidoglycan modifications include O-acetylation and N-glycolylation of muramic acid and N-deacetylation of N-acetylglucosamine (Davis et al. 2011). Modification of mycobacterial peptidoglycan by N-glycolylation prevents the catalytic activity of lysozyme (Raymond et al. 2005). Additionally, mycobacterial peptidoglycan is modified by amidation for unknown reasons.
Here, we have investigated the role of amidated peptidoglycan in Mycobacterium sp in modulating the innate immune response. We isolated amidated peptidoglycan from Mycobacterium sp and non-amidated peptidoglycan from Escherichia coli. We made a comparative analysis of the cytokine response produced on stimulation of innate immune cells by peptidoglycan from E. Coli and Mycobacterium sp. Macrophages and whole blood were treated with peptidoglycan and the cytokines secreted into spent medium and plasma respectively were analyzed using ELISA. Our results show that peptidoglycan from Mycobacterium sp is less effective in stimulating innate immune cells to produce cytokines. This intrinsic modulation of the cytokine response suggests that mycobacteria modify their peptidoglycan by amidation to evade innate immune response.