Vural Özdemir Ph.D.
Sanjeeva Srivastava Ph.D.
 Bharat B. Chattoo, Ph.D.
Bharat B. Chattoo, Ph.D.
Professor, Faculty of Science M.S.University of Baroda, India
Biology of plant infection by Magnaporthe oryzae
The rice blast disease caused by the ascomycetous fungus Magnaporthe oryzae is a major constraint in rice production. Rice-M.oryzae is also emerging as a good model patho-system to investigate how the fungus invades and propagates within the host. Identification and characterisation of genes critical for fungal pathogenesis provides opportunities to explore their use as possible targets for development of strategies for combating fungal infection and to better understand the complex process of host-pathogen interaction.
We have used insertional mutagenesis and RNAi based approaches to identify pathogenesis related genes in this fungus. A large number of mutants were isolated using Agrobacterium tumefaciens mediated transformation (ATMT). Characterisation of several interesting mutants is in progress. We have identified a novel gene, MGA1, required for the development of appressoria. The mutant mga1 is unable to infect and is impaired in glycogen and lipid mobilization required for appressorium development. The glycerol content in the mycelia of the mutant was significantly lower as compared to wild type and it was unable to tolerate hyperosmotic stress. A novel ABC transporter was identified in this fungus. The abc4 mutant did not form functional appressoria, was non-pathogenic and showed increased sensitivity to certain antifungal molecules implying the role of ABC4 in multidrug resistance (MDR). Another mutant MoSUMO (MGG_05737) was isolated using a Split Marker technique; the mutant showed defects in growth, germination and infection. Immuno-fluorescence microscopy revealed that MoSumo is localized to septa in mycelia and nucleus as well as septa in spores. Two Dimensional Gel Electrophoresis showed differences in patterns of protein expression between Wild Type B157 and MoΔSumo mutant. We also isolated and charaterised mutants in MoALR2 (MGG_08843) and MoMNR2 (MGG_09884). Our results indicate that both MoALR2 and MoMNR2 are Mg2+ transporters, and the reduction in the levels of CorA transporters caused defects in surface hydrophobicity, cell wall stress tolerance, sporulation, appressorium formation and infection are mediated through changes in the key signaling cascades in the knock-down transformants. (Work supported by the Department of Biotechnology, Government of India)
 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
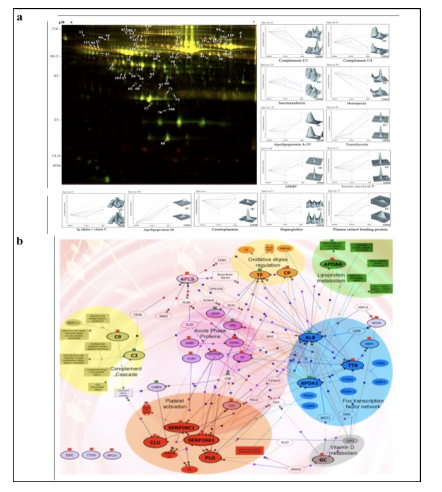
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Leland H. Hartwell Ph.D.
Leland H. Hartwell Ph.D.
2001 Nobel Laureate, Physiology & Medicine
Dr. Lee Hartwell received the 2001 Nobel Prize in Physiology / Medicine for his discovery of protein molecules that control the division of cells. He was the President and Director of the Fred Hutchinson Cancer Research Center in Seattle, Washington before moving to Arizona State University’s Center for Sustainable Health.
Dr. Hartwell is also adjunct faculty at Amrita University. He spoke to the delegates at Bioquest from his office in the US, over Amrita’s e-learning platform A-View. Given below are excerpts from his address.
I would like to address the young people in the audience. I know that many of you may have come to this meeting wondering, “How can I become a successful scientist? How can I prepare myself to make a contribution in this world?”
These questions are interesting to me also.
Believe it or not, I am still trying to be a successful scientist. That may surprise you since you probably think that a Nobel laureate must have found the answers. But the problem is that the answers to these questions change with time and the answers are different today than what they were when I began my career fifty years ago. The strategy of the 1960’s doesn’t work so well anymore. What is different now?
First, what we know now is much more. For example, by 1970, no genes from any organisms were sequenced. In 2013, we have the complete sequence of the human genome. Second, not only do we know much more today, accessing that knowledge is easy. Third, obtaining new information is much faster today.
Our rich understanding of science and technology is now needed to solve many serious problems. The human population has reached the size where we are utilizing all available resource of the planet. We are utilizing all of the agricultural land, all of the water, all of the forest and fishing resources. We are also polluting the planet that we live on.
We are polluting the land with fertilizers and pesticides; the oceans with acids and the atmosphere with carbon dioxide. We are using up top soil and ground water, thereby reducing our capacity to feed ourselves. We are using up petroleum, the energy source that our entire economy is dependent on. These are problems we were largely unaware of, fifty years ago. But these are problems that must be solved in your life times.
The big question facing your generation is, how can human beings live sustainably on planet earth. Your two broad goals on sustainability are 1) leave the planet as you first found it for your future generations; don’t use up the resources and don’t pollute the planet 2) everyone deserves to have an equal share of the earth’s resources.
Income strongly determines one’s opportunities in life. Many poor people succumb to chronic diseases and unhealthy environments. This inequality undermines our ability to live sustainably. We can’t ask the poor to leave the planet as they found it if they can’t support their families. Education, healthcare, employment are essential to having a sustainable society.
How can we be a successful scientist in 2013?
1. First choose a problem to solve
2. Ask questions to understand why it is not solved
3. Collaborate with those who can help
4. Develop a solution that works in the real worldChronic diseases are our major burden and this burden will get worse. Heart disease, diabetes, cancer, dementia and other diseases. The good news is that the chronic diseases are largely preventable and more easily curable if detected early. One question that attracts me is how can we detect disease earlier when it can be more easily cured?
Can we use our increasing knowledge in molecular biology to identify biomarkers for early disease detection?
We need to collaborate very closely with clinicians who care for patients to find out exactly where they need help.
I think if we apply our technology to important clinical questions we will actually save medical expenditure and be well on our way to making a great contribution to society.



 D. Narasimha Rao, Ph.D.
D. Narasimha Rao, Ph.D.
Professor, Dept of Biochemistry, Indian Institute of Science, Bangalore, India
Genomics of Restriction-Modification Systems
Restriction endonucleases occur ubiquitously among procaryotic organisms. Up to 1% of the genome of procaryotic organisms is taken up by the genes for these enzymes. Their principal biological function is the protection of the host genome against foreign DNA, in particular bacteriophage DNA. Restriction-modification (R-M) systems are composed of pairs of opposing enzyme activities: an endonuclease and a DNA methyltransferase (MTase). The endonucleases recognise specific sequences and catalyse cleavage of double-stranded DNA. The modification MTases catalyse the addition of a methyl group to one nucleotide in each strand of the recognition sequence using S-adenosyl-L-methionine (AdoMet) as the methyl group donor. Based on their molecular structure, sequence recognition, cleavage position and cofactor requirements, R-M systems are generally classified into three groups. In general R-M systems restrict unmodified DNA, but there are other systems that specifically recognise and cut modified DNA. More than 3500 restriction enzymes have been discovered so far. With the identification and sequencing of a number of R-M systems from bacterial genomes, an increasing number of these have been found that do not seem to fit into the conventional classification.
It is well documented that restriction enzyme genes always lie close to their cognate methyltransferase genes. Analysis of the bacterial and archaeal genome sequences shows that MTase genes are more common than one would have expected on the basis of previous biochemical screening. Frequently, they clearly form part of a R-M system, because the adjacent open reading frames (ORFs) show similarity to known restriction enzyme genes. Very often, though, the adjacent ORFs have no homologs in the GenBank and become candidates either for restriction enzymes with novel specificities or for new examples of previously uncloned specificities. Sequence-dependent modification and restriction forms the foundation of defense against foreign DNAs and thus RM systems may serve as a tool of defense for bacterial cells. RM systems however, sometimes behave as discrete units of life, and any threat to their maintenance, such as a challenge by a competing genetic element can lead to cell death through restriction breakage in the genome, thus providing these systems with a competitive advantage. The RM systems can behave as mobile-genetic elements and have undergone extensive horizontal transfer between genomes causing genome rearrangements. The capacity of RM systems to act as selfish, mobile genetic elements may underlie the structure and function of RM enzymes.
The similarities and differences in the different mechanisms used by restriction enzymes will be discussed. Although it is not clear whether the majority of R-M systems are required for the maintenance of the integrity of the genome or whether they are spreading as selfish genetic elements, they are key players in the “genomic metabolism” of procaryotic organisms. As such they deserve the attention of biologists in general. Finally, restriction enzymes are the work horses of molecular biology. Understanding their enzymology will be advantageous to those who use these enzymes, and essential for those who are devoted to the ambitious goal of changing the properties of these enzymes, and thereby make them even more useful.


H S M Kort, J-W J Lammers, S N W Vorrink, T Troosters
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a disabling airway disease with variable extrapulmonary effects that may contribute to disease severity in individual patients (Rabe et al. 2007). The world health organization predicts that COPD will become the third leading cause of death worldwide by 2030. Patients with COPD demonstrate reduced levels of spontaneous daily physical activity (DPA) compared with healthy controls (Pitta et al. 2005). This results in a higher risk of hospital admission and shorter survival (Pitta et al. 2006). Pulmonary rehabilitation can help to improve the DPA level, however, obtained benefits decline after 1–2 years (Foglio et al. 2007).
Purpose
In order to maintain DPA in COPD patients after rehabilitation, we developed a mobile phone application. This application measures DPA as steps per day, measured by the accelerometer of the smartphone, and shows the information to the patient via the display of the mobile phone. A physiotherapist can monitor the patient via a secure website where DPA measurements are visible for all patients. Here, DPA goals can be adjusted and text messages sent.
Method
Three pilot studies were performed with healthy students and COPD patients to test the application for usability, user friendliness and reliability with questionnaires and focus groups. Subjects also wore a validated accelerometer. For the Randomized Controlled Trial (RCT) 140 COPD patients will be recruited in Dutch physiotherapy practises. They will be randomised in an intervention group that receives the smartphone for 6 months and a control group. Measurements include lungfunction, dyspnea, and exercise capacity and are held at 0, 3, 6 and 12 months.
Results and Discussion
The application was found to be useful, easy to learn and use. Subjects had no problems with health care professionals seeing information on their physical activity performance. They do find it important to be able to determine who can see the information. Correlations between the accelerometer and the measurements on DPA of the smartphone for steps per hour were 0.69 and 0.70 for pilot studies 1 (students) and 2 (COPD patients) respectively. The version of the application in pilot study 3 contained an error, which made correlations with the accelerometer unusable. The RCT study is now being executed.
 Vural Özdemir, MD, Ph.D., DABCP
Vural Özdemir, MD, Ph.D., DABCP
Co-Founder, DELSA Global, Seattle, WA, USA
Crowd-Funded Micro-Grants to Link Biotechnology and “Big Data” R&D to Life Sciences Innovation in India
Vural Özdemir, MD, PhD, DABCP1,2*
- Data-Enabled Life Sciences Alliance International (DELSA Global), Seattle, WA 98101, USA;
- Faculty of Management and Medicine, McGill University, Canada;
ABSTRACT
Aims: This presentation proposes two innovative funding solutions for linking biotechnology and “Big Data” R&D in India with artisan small scale discovery science, and ultimately, with knowledge-based innovation:
- crowd-funded micro-grants, and
- citizen philanthropy
These two concepts are new, and inter-related, and can be game changing to achieve the vision of biotechnology innovation in India, and help bridge local innovation with global science.
Background and Context: Biomedical science in the 21(st) century is embedded in, and draws from, a digital commons and “Big Data” created by high-throughput Omics technologies such as genomics. Classic Edisonian metaphors of science and scientists (i.e., “the lone genius” or other narrow definitions of expertise) are ill equipped to harness the vast promises of the 21(st) century digital commons. Moreover, in medicine and life sciences, experts often under-appreciate the important contributions made by citizen scholars and lead users of innovations to design innovative products and co-create new knowledge. We believe there are a large number of users waiting to be mobilized so as to engage with Big Data as citizen scientists-only if some funding were available. Yet many of these scholars may not meet the meta-criteria used to judge expertise, such as a track record in obtaining large research grants or a traditional academic curriculum vitae. This presentation will describe a novel idea and action framework: micro-grants, each worth $1000, for genomics and Big Data. Though a relatively small amount at first glance, this far exceeds the annual income of the “bottom one billion” – the 1.4 billion people living below the extreme poverty level defined by the World Bank ($1.25/day).
We will present two types of micro-grants. Type 1 micro-grants can be awarded through established funding agencies and philanthropies that create micro-granting programs to fund a broad and highly diverse array of small artisan labs and citizen scholars to connect genomics and Big Data with new models of discovery such as open user innovation. Type 2 micro-grants can be funded by existing or new science observatories and citizen think tanks through crowd-funding mechanisms described herein. Type 2 micro-grants would also facilitate global health diplomacy by co-creating crowd-funded micro-granting programs across nation-states in regions facing political and financial instability, while sharing similar disease burdens, therapeutics, and diagnostic needs. We report the creation of ten Type 2 micro-grants for citizen science and artisan labs to be administered by the nonprofit Data-Enabled Life Sciences Alliance International (DELSA Global, Seattle: http://www.delsaglobal.org). Our hope is that these micro-grants will spur novel forms of disruptive innovation and life sciences translation by artisan scientists and citizen scholars alike.
Address Correspondence to:
Vural Özdemir, MD, PhD, DABCP
Senior Scholar and Associate Professor
Faculty of Management and Medicine, McGill University
1001 Sherbrooke Street West
Montreal, Canada H3A 1G5
Email: vural.ozdemir@alumni.utoronto.ca




Tejaswini Subbannayya, Nandini A. Sahasrabuddhe, Arivusudar Marimuthu, Santosh Renuse, Gajanan Sathe, Srinivas M. Srikanth, Mustafa A. Barbhuiya, Bipin Nair, Juan Carlos Roa, Rafael Guerrero-Preston, H. C. Harsha, David Sidransky, Akhilesh Pandey, T. S. Keshava Prasad and Aditi Chatterjee
Proteomic profiling of gallbladder cancer secretome – a source for circulatory biomarker discovery
Gallbladder cancer (GBC) is the fifth most common cancer of the gastrointestinal tract and one of the common malignancies that occur in the biliary tract (Misra et al. 2006; Lazcano-Ponce et al. 2001). It has a poor prognosis with survival of less than 5 years in 90% of the cases (Misra et al. 2003). The etiology is ill-defined. Several risk factors have been reported including cholelithiasis, obesity, female gender and exposure to carcinogens (Eslick 2010; Kumar et al. 2006). Poor prognosis in GBC is mainly due to late presentation of the disease and lack of reliable biomarkers for early diagnosis. This emphasizes the need to identify and characterize cancer biomarkers to aid in the diagnosis and prognosis of GBC. Secreted proteins are an important class of molecules which can be detected in body fluids and has been targeted for biomarker discovery. There are challenges faced in the proteomic interrogation of body fluids especially plasma such as low abundance of tumor secreted proteins, high complexity and high abundance of other proteins that are not released by the tumor cells (Tonack et al. 2009). Profiling of conditioned media from the cancer cell lines can be used as an alternate means to identify secreted proteins from tumor cells (Kashyap et al. 2010; Marimuthu et al. 2012). We analyzed the invasive property of 7 GBC cell lines (SNU-308, G-415, GB-d1, TGBC2TKB, TGBC24TKB, OCUG-1 and NOZ). Four cell lines were selected for analysis of the cancer secretome based on the invasive property of the cells. We employed isobaric tags for relative and absolute quantitation (iTRAQ) labeling technology coupled with high resolution mass spectrometry to identify and characterize secretome from the panel of 4GBC cancer cells mentioned above. In total, we have identified around 2,000 proteins of which 175 were secreted at differential abundance across all the four cell lines. This secretome analysis will act as a reservoir of candidate biomarkers. Currently, we are investigating and validating these candidate markers from GBC cell secretome. Through this study, we have shown mass spectrometry-based quantitative proteomic analysis as a robust approach to investigate secreted proteins in cancer cells.