 Gillian Murphy, Ph.D.
Gillian Murphy, Ph.D.
Professor, Department of Oncology, University of Cambridge, UK
A novel strategy for targeting metalloproteinases in cancer
Epithelial tumours evolve in a multi-step manner, involving both inflammatory and mesenchymal cells. Although intrinsic factors drive malignant progression, the influence of the micro-environment of neoplastic cells is a major feature of tumorigenesis. Extracellular proteinases, notably the metalloproteinases, are key players in the regulation of this cellular environment, acting as major effectors of both cell-cell and cell-extracellular matrix (ECM) interactions. They are involved in modifying ECM integrity, growth factor availability and the function of cell surface signalling systems, with consequent effects on cellular differentiation, proliferation and apoptosis.This has made metalloproteinases important targets for therapeutic interventions in cancer and small molecule inhibitors focussed on chelation of the active site zinc and binding within the immediate active site pocket were developed. These were not successful in early clinical trials due to the relative lack of specificity and precise knowledge of the target proteinase(s) in specific cancers. We can now appreciate that it is essential that we understand the relative roles of the different enzymes (of which there are over 60) in terms of their pro and anti tumour activity and their precise sites of expression The next generations of metalloproteinase inhibitors need the added specificity that might be gained from an understanding of the structure of individual active sites and the role of extra catalytic domains in substrate binding and other aspects of their biology. We have prepared scFv antibodies to the extra catalytic domains of two membrane metalloproteinases, MMP-14 and ADAM17, that play key roles in the tumour microenvironment. Our rationale and experiences with these agents will be presented in more detail.

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
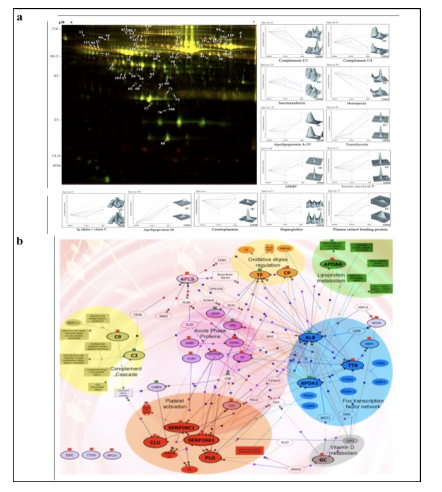
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 D. Narasimha Rao, Ph.D.
D. Narasimha Rao, Ph.D.
Professor, Dept of Biochemistry, Indian Institute of Science, Bangalore, India
Genomics of Restriction-Modification Systems
Restriction endonucleases occur ubiquitously among procaryotic organisms. Up to 1% of the genome of procaryotic organisms is taken up by the genes for these enzymes. Their principal biological function is the protection of the host genome against foreign DNA, in particular bacteriophage DNA. Restriction-modification (R-M) systems are composed of pairs of opposing enzyme activities: an endonuclease and a DNA methyltransferase (MTase). The endonucleases recognise specific sequences and catalyse cleavage of double-stranded DNA. The modification MTases catalyse the addition of a methyl group to one nucleotide in each strand of the recognition sequence using S-adenosyl-L-methionine (AdoMet) as the methyl group donor. Based on their molecular structure, sequence recognition, cleavage position and cofactor requirements, R-M systems are generally classified into three groups. In general R-M systems restrict unmodified DNA, but there are other systems that specifically recognise and cut modified DNA. More than 3500 restriction enzymes have been discovered so far. With the identification and sequencing of a number of R-M systems from bacterial genomes, an increasing number of these have been found that do not seem to fit into the conventional classification.
It is well documented that restriction enzyme genes always lie close to their cognate methyltransferase genes. Analysis of the bacterial and archaeal genome sequences shows that MTase genes are more common than one would have expected on the basis of previous biochemical screening. Frequently, they clearly form part of a R-M system, because the adjacent open reading frames (ORFs) show similarity to known restriction enzyme genes. Very often, though, the adjacent ORFs have no homologs in the GenBank and become candidates either for restriction enzymes with novel specificities or for new examples of previously uncloned specificities. Sequence-dependent modification and restriction forms the foundation of defense against foreign DNAs and thus RM systems may serve as a tool of defense for bacterial cells. RM systems however, sometimes behave as discrete units of life, and any threat to their maintenance, such as a challenge by a competing genetic element can lead to cell death through restriction breakage in the genome, thus providing these systems with a competitive advantage. The RM systems can behave as mobile-genetic elements and have undergone extensive horizontal transfer between genomes causing genome rearrangements. The capacity of RM systems to act as selfish, mobile genetic elements may underlie the structure and function of RM enzymes.
The similarities and differences in the different mechanisms used by restriction enzymes will be discussed. Although it is not clear whether the majority of R-M systems are required for the maintenance of the integrity of the genome or whether they are spreading as selfish genetic elements, they are key players in the “genomic metabolism” of procaryotic organisms. As such they deserve the attention of biologists in general. Finally, restriction enzymes are the work horses of molecular biology. Understanding their enzymology will be advantageous to those who use these enzymes, and essential for those who are devoted to the ambitious goal of changing the properties of these enzymes, and thereby make them even more useful.

 Karmeshu, Ph.D.
Karmeshu, Ph.D.
Dean & Professor, School of Computer & Systems Sciences & School of Computational & Integrative Sciences, Jawaharlal Nehru University, India.
Interspike Interval Distribution of Neuronal Model with distributed delay: Emergence of unimodal, bimodal and Power law
The study of interspike interval distribution of spiking neurons is a key issue in the field of computational neuroscience. A wide range of spiking patterns display unimodal, bimodal ISI patterns including power law behavior. A challenging problem is to understand the biophysical mechanism which can generate the empirically observed patterns. A neuronal model with distributed delay (NMDD) is proposed and is formulated as an integro-stochastic differential equation which corresponds to a non-markovian process. The widely studied IF and LIF models become special cases of this model. The NMDD brings out some interesting features when excitatory rates are close to inhibitory rates rendering the drift close to zero. It is interesting that NMDD model with gamma type memory kernel can also account for bimodal ISI pattern. The mean delay of the memory kernels plays a significant role in bringing out the transition from unimodal to bimodal ISI distribution. It is interesting to note that when a collection of neurons group together and fire together, the ISI distribution exhibits power law.

Syed Salman Lateef and Vinayak A K
Development of Supercritical Fluid Chromatography methods for the replacement of existing USP Normal phase liquid chromatography methods
Normal phase liquid chromatography methods often have long run times and involve environmentally toxic/costly solvents. Supercritical chromatography methods on the other hand are faster, inexpensive, and eco-friendly. The low viscous supercritical carbon dioxide operates at high flow rates compared to LC without losing separation efficiency. In this work, SFC methods are developed to replace three United States Pharmacopeial (USP) normal phase achiral methods – prednisolone, tolazamide and cholecalciferol. System suitability parameters of the normal phase method are compared against the SFC method. Precision, linearity and robustness of the new SFC methods are demonstrated. SFC methods were found to be cost effective in terms of analysis time and solvent savings. The SFC method does not require purchase and disposal of expensive environmentally hazardous chemicals. Hence, the newly developed SFC method provides a faster and safer solution.
 Sudarslal S, Ph.D.
Sudarslal S, Ph.D.
Associate Professor, School of Biotechnology, Amrita University
Electrospray ionization ion trap mass spectrometry for cyclic peptide characterization
There has been considerable interest in the isolation and structural characterization of bioactive peptides produced by bacteria and fungi. Most of the peptides are cyclic depsipeptides characterized by the presence of lactone linkages and β-hydroxy fatty acids. Occurrence of microheterogeneity is another remarkable property of these peptides. Even if tandem mass spectrometers are good analytical tools to structurally characterize peptides and proteins, sequence analysis of cyclic peptides is often ambiguous due to the random ring opening of the peptides and subsequent generation of a set of linear precursor ions with the same m/z. Here we report combined use of chemical derivatization and multistage fragmentation capability of ion trap mass spectrometers to determine primary sequences of a series of closely related cyclic peptides.


Ravindra Gudihal, Suresh Babu C V
Bioanalytical Characterization of Therapeutic Proteins
The characterization of therapeutic proteins such as monoclonal antibody (mAb) during different stages of manufacturing is crucial for timely and successful product release. Regulatory agencies require a variety of analytical technologies for comprehensive and efficient protein analysis. Electrophoresis-based techniques and liquid chromatography (LC) either standalone or coupled to mass spectrometry (MS) are at the forefront for the in-depth analysis of protein purity, isoforms, stability, aggregation, posttranslational modifications, PEGylation, etc. In this presentation, a combination of various chromatographic and electrophoretic techniques such as liquid-phase isoelectric focusing, microfluidic and capillary-based electrophoresis (CE), liquid chromatography (LC) and combinations of those with mass spectrometry techniques will be discussed. We present a workflow based approach to the analysis of therapeutic proteins. In successive steps critical parameters like purity, accurate mass, aggregation, peptide sequence, glycopeptide and glycan analysis are analyzed. In brief, the workflow involved proteolytic digestion of therapeutic protein for peptide mapping, N-Glycanase and chemical labeling reaction for glycan analysis, liquid-phase isoelectric focusing for enrichment of charge variants followed by a very detailed analysis using state of the art methods such as CE-MS and LC-MS. For the analysis of glycans, we use combinations of CE-MS and LC-MS to highlight the sweet spots of these techniques. CE-MS is found to be more useful in analysis of highly sialylated glycans (charged glycans) while nano LC-MS seems to be better adapted for analysis of neutral glycans. These two techniques can be used to get complementary data to profile all the glycans present in a given protein. In addition, microfluidic electrophoresis was used as a QC tool in initial screening for product purity, analysis of papain digestion fragments of mAb, protein PEGylation products, etc. The described workflow involves multiple platforms, provides an end to end solution for comprehensive protein characterization and aims at reducing the total product development time.