 Krishnakumar Menon, Ph.D.
Krishnakumar Menon, Ph.D.
Associate Professor, Centre for Nanosciences & Molecular Medicine, Amrita University, Kochi, India
A Far-Western Clinical Proteomics Approach to Detect Molecules of Clinical and Pathological Significance in the Neurodegenerative Disease Multiple Sclerosis.
Multiple Sclerosis (MS), an autoimmune neurodegenerative disorder of the central nervous system. The disease affects young adults at their prime age leading to severe debilitation over several years. Despite advances in MS research, the cause of the disease remains elusive. Thus, our objective is to identify novel molecules of pathological and diagnostic significance important in the understanding, early diagnosis and treatment of MS. Biological fluids such as cerebrospinal fluid (CSF), that bathe the brain serve as a potential source for the identification of pathologically significant autoantibody reactivity in MS. In this regard, we report the development of an unbiased clinical proteomics approach for the detection of reactive CSF molecules that target brain proteins from patients with MS. Proteins of myelin and myelin-axolemmal complexes were separated by two-dimensional gel electrophoresis, blotted onto membranes and probed separately with biotinylated unprocessed CSF samples. Protein spots that reacted specifically to MS-CSF’s were further analyzed by matrix assisted laser desorption ionization-time-of-flight time-of-flight mass spectrometry. In addition to previously reported proteins found in MS, we have identified several additional molecules involved in mitochondrial and energy metabolism, myelin gene expression and/or cytoskeletal organization. Among these identified molecules, the cellular expression pattern of collapsin response mediator protein-2 and ubiquitin carboxy-terminal hydrolase L1 were investigated in human chronic-active MS lesions by immunohistochemistry. The observation that in multiple sclerosis lesions phosphorylated collapsin response mediator protein-2 was increased, whereas Ubiquitin carboxy-terminal hydrolase L1 was down-regulated, not only highlights the importance of these molecules in the pathology of this disease, but also illustrates the use of our approach in attempting to decipher the complex pathological processes leading to multiple sclerosis and other neurodegenerative diseases. Further, we show that in clinicaly isolated syndrome (CIS), we could identify important molecules that could serve as an early diagnostic biomarker in MS.

 Egidio D’Angelo, MD, Ph.D.
Egidio D’Angelo, MD, Ph.D.
Full Professor of Physiology & Director, Brain Connectivity Center, University of Pavia, Italy
Realistic modeling: new insight into the functions of the cerebellar network
Realistic modeling is an approach based on the careful reconstruction of neurons synapses starting from biological details at the molecular and cellular level. This technique, combined with the connection topologies derived from histological measurements, allows the reconstruction of precise neuronal networks. Finally, the advent of specific software platforms (PYTHON-NEURON) and of super-computers allows large-scale network simulation to be performed in reasonable time. This approach inverts the logics of older theoretical models, which anticipated an intuition on how the network might work. In realistic modeling, network properties “emerge” from the numerous biological properties embedded into the model.
This approach is illustrated here through an outstanding application of realistic modeling to the cerebellar cortex network. The neurons (over 105) are reproduced at a high level of detail generating non-linear network effects like population oscillations and resonance, phase-reset, bursting, rebounds, short-term and long-term plasticity, spatiotemporal redistrbution of input patterns. The model is currently being used in the context of he HUMAN BRAIN PROJECT to investigate the cerebellar network function.
Correspondence should be addressed to
Dr. EgidioD’Angelo,
Laboratory of Neurophysiology
Via Forlanini 6, 27100 Pavia, Italy
Phone: 0039 (0) 382 987606
Fax: 0039 (0) 382 987527
dangelo@unipv.it
Acknowledgments
This work was supported by grants from European Union to ED (CEREBNET FP7-ITN238686, REALNET FP7-ICT270434) and by grants from the Italian Ministry of Health to ED (RF-2009-1475845).

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
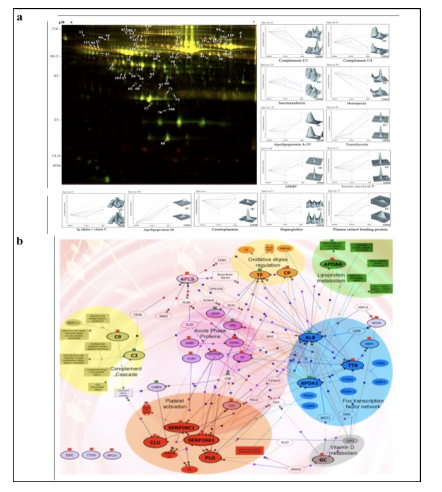
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)



Manjunath Joshi, Apoorva Lad, Bharat Prasad Alevoor, Aswath Balakrishnan, Lingadakai Ramachandra and Kapaettu Satyamoorthy
Pathological conditions during Type 2 Diabetes (T2D) are associated with elevated risk for common community acquired infections due to poor glycemic control. Multiple studies have indicated specific defects in innate and adaptive immune function in diabetic subjects. Neutrophils play an important role in eliminating pathogens as an active constituent of innate immune system. Apart from canonically known phagocytosis mechanism, neutrophils are endowed with a unique ability to produce extracellular traps (NETs) to kill pathogens by expelling DNA coated with bactericidal proteins and histone. NETosis is stimulated by diverse bacteria and their products, fungi, protozoans, cytokines, phorbol esters and by activated platelets. Considering deregulation of metabolic and immune response pathways during pathological state of diabetes and NETosis as a potential mechanism for killing bacteria, we therefore, investigated whether hyperglycemic conditions modulate formation of neutrophil NETs and attempted to identify underlying immunoregulatory mechanisms. Freshly isolated neutrophils from normal individuals were cultured in absence or presence of high glucose (different concentrations) for 24 hours and activated with either LPS (2 mg/ml) or PMA (20 ng/ml) or IL-6 (20 ng/ml) for 3 hours. NETs were visualized and quantified by addition of DNA binding dye SYTOX green using fluorescence microscope and fluorimetry. NETs were quantified in Normal and diabetic subjects. Serum IL-6 levels were measured using ELISA technique. NETs bound elasatse were quantified in normal and diabetic subjects in presence or absence of DNase. Bacterial killing assays were performed upon infecting E.coli with activated neutrophils from normal and diabetic subjects. Microscopy and fluorimetry analysis suggested dramatic impairment in NETs formation under high glucose conditions. Extracellular DNA lattices formed in hyperglycemic conditions were short lived and unstable leading to rapid disintegration. Subsequent, time course experiments showed that NETs production was delayed in hyperglycemic conditions. To validate our findings more closely to clinical conditions, we investigated the neutrophil activation and NETs formation in diabetic patients. Upon stimulation with LPS for three hours, neutrophils from diabetic subjects responded weakly to LPS and lesser NETs were formed; whereas, neutrophils from normal individuals showed robust release of NETs. In few patients we found short and imperfect NETs in basal conditions suggesting constitutive activation of neutrophils in diabetic subjects. Interestingly, NETs bound elastase activity was reduced in diabetes subjects when compared to non-diabetic individuals, indicating a dysfunction of one of the important protein component of NETs during diabetes. Neutrophils from diabetic subjects released higher levels of IL-6 without any stimulation suggesting an existence of constitutively activated pro-inflammatory state. IL-6 induced NETs formation and was abrogated by high glucose. Weobserved that glycolysis inhibitor 2-DG resensitize the high glucose attenuated LPS and IL-6 induced NETs. a) NETs are influenced by glucose homeostasis, b) IL-6 as potent inducer of energy dependent NETs formation and c) hyperglycemia mimics a state of constitutively active pro-inflammatory condition in neutrophils leading to reduced response to external stimuli making diabetic subjects susceptible for infections.
 Sudarslal S, Ph.D.
Sudarslal S, Ph.D.
Associate Professor, School of Biotechnology, Amrita University
Electrospray ionization ion trap mass spectrometry for cyclic peptide characterization
There has been considerable interest in the isolation and structural characterization of bioactive peptides produced by bacteria and fungi. Most of the peptides are cyclic depsipeptides characterized by the presence of lactone linkages and β-hydroxy fatty acids. Occurrence of microheterogeneity is another remarkable property of these peptides. Even if tandem mass spectrometers are good analytical tools to structurally characterize peptides and proteins, sequence analysis of cyclic peptides is often ambiguous due to the random ring opening of the peptides and subsequent generation of a set of linear precursor ions with the same m/z. Here we report combined use of chemical derivatization and multistage fragmentation capability of ion trap mass spectrometers to determine primary sequences of a series of closely related cyclic peptides.



Ravindra Gudihal, Suresh Babu C V
Bioanalytical Characterization of Therapeutic Proteins
The characterization of therapeutic proteins such as monoclonal antibody (mAb) during different stages of manufacturing is crucial for timely and successful product release. Regulatory agencies require a variety of analytical technologies for comprehensive and efficient protein analysis. Electrophoresis-based techniques and liquid chromatography (LC) either standalone or coupled to mass spectrometry (MS) are at the forefront for the in-depth analysis of protein purity, isoforms, stability, aggregation, posttranslational modifications, PEGylation, etc. In this presentation, a combination of various chromatographic and electrophoretic techniques such as liquid-phase isoelectric focusing, microfluidic and capillary-based electrophoresis (CE), liquid chromatography (LC) and combinations of those with mass spectrometry techniques will be discussed. We present a workflow based approach to the analysis of therapeutic proteins. In successive steps critical parameters like purity, accurate mass, aggregation, peptide sequence, glycopeptide and glycan analysis are analyzed. In brief, the workflow involved proteolytic digestion of therapeutic protein for peptide mapping, N-Glycanase and chemical labeling reaction for glycan analysis, liquid-phase isoelectric focusing for enrichment of charge variants followed by a very detailed analysis using state of the art methods such as CE-MS and LC-MS. For the analysis of glycans, we use combinations of CE-MS and LC-MS to highlight the sweet spots of these techniques. CE-MS is found to be more useful in analysis of highly sialylated glycans (charged glycans) while nano LC-MS seems to be better adapted for analysis of neutral glycans. These two techniques can be used to get complementary data to profile all the glycans present in a given protein. In addition, microfluidic electrophoresis was used as a QC tool in initial screening for product purity, analysis of papain digestion fragments of mAb, protein PEGylation products, etc. The described workflow involves multiple platforms, provides an end to end solution for comprehensive protein characterization and aims at reducing the total product development time.