 Egidio D’Angelo, MD, Ph.D.
Egidio D’Angelo, MD, Ph.D.
Full Professor of Physiology & Director, Brain Connectivity Center, University of Pavia, Italy
Realistic modeling: new insight into the functions of the cerebellar network
Realistic modeling is an approach based on the careful reconstruction of neurons synapses starting from biological details at the molecular and cellular level. This technique, combined with the connection topologies derived from histological measurements, allows the reconstruction of precise neuronal networks. Finally, the advent of specific software platforms (PYTHON-NEURON) and of super-computers allows large-scale network simulation to be performed in reasonable time. This approach inverts the logics of older theoretical models, which anticipated an intuition on how the network might work. In realistic modeling, network properties “emerge” from the numerous biological properties embedded into the model.
This approach is illustrated here through an outstanding application of realistic modeling to the cerebellar cortex network. The neurons (over 105) are reproduced at a high level of detail generating non-linear network effects like population oscillations and resonance, phase-reset, bursting, rebounds, short-term and long-term plasticity, spatiotemporal redistrbution of input patterns. The model is currently being used in the context of he HUMAN BRAIN PROJECT to investigate the cerebellar network function.
Correspondence should be addressed to
Dr. EgidioD’Angelo,
Laboratory of Neurophysiology
Via Forlanini 6, 27100 Pavia, Italy
Phone: 0039 (0) 382 987606
Fax: 0039 (0) 382 987527
dangelo@unipv.it
Acknowledgments
This work was supported by grants from European Union to ED (CEREBNET FP7-ITN238686, REALNET FP7-ICT270434) and by grants from the Italian Ministry of Health to ED (RF-2009-1475845).

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
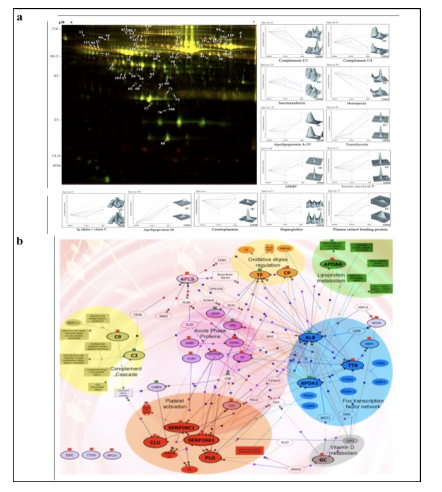
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Sharmila Mande, Ph.D.
Sharmila Mande, Ph.D.
Principal Scientist and Head, Bio Sciences R&D, TCS Innovation Labs, Pune
Gut microbiome and health: Moving towards the new era of translational medicine
The microbes inhabiting our body outnumber our own cells by a factor of 10. The genomes of these microbes, called the ‘second genome’ are therefore expected to have great influence on our health and well being. The emerging field of metagenomics is rapidly becoming the method of choice for studying the microbial community (called microbiomes) present in various parts of the human body. Recent studies have implicated the role of gut microbiomes in several diseases and disorders. Studies have indicated gut microbiome’s role in nutrient absorption, immuno-modulation motor-response, and other key physiological processes. However, our understanding of the role of gut microbiota in malnutrition is currently incomplete. Exploration of these aspects are likely to help in understanding the microbial basis for several physiological disorders associated with malnutrition (eg, increased susceptibility to diarrhoeal pathogens) and may finally aid in devising appropriate probiotic strategies addressing this menace. A metagenomic approach was employed for analysing the differences between gut microbial communities obtained from malnourished and healthy children. Results of the analysis using TCS’ ‘Metagenomic Analysis Platform’ were discussed in detail during my talk.

Aswath Balakrishnan, Kapaettu Satyamoorthy and Manjunath B Joshi
Introduction
Insulin resistance is a hall mark of metabolic disorders such as diabetes. Reduced insulin response in vasculature leads to disruption of IR/Akt/eNOS signaling pathway resulting in vasoconstriction and subsequently to cardiovascular diseases. Recent studies have demonstrated that inflammatory regulator interleukin-6 (IL-6), as one of the potential mediators that can link chronic inflammation with insulin resistance. Accumulating evidences suggest a significant role of epigenetic mechanisms such as DNA methylation in progression of metabolic disorders. Hence the present study aimed to understand the role of epigenetic mechanisms involved during IL-6 induced vascular insulin resistance and its consequences in cardiovascular diseases.
Materials and Methods
Human umbilical vein endothelial cells (HUVEC) and Human dermal microvascular endothelial cells (HDMEC) were used for this study. Endothelial cells were treated in presence or absence of IL-6 (20ng/ml) for 36 hours and followed by insulin (100nM) stimulation for 15 minutes. Using immunoblotting, cell lysates were stained for phosphor- and total Akt levels to measure insulin resistance. To investigate changes in DNA methylation, cells were treated with or without neutrophil conditioned medium (NCM) as a physiological source of inflammation or IL-6 (at various concentrations) for 36 hours. Genomic DNA was processed for HPLC analysis for methyl cytosine content and cell lysates were analyzed for DNMT1 (DNA (cytosine-5)-methyltransferase 1) and DNMT3A (DNA (cytosine-5)-methyltransferase 3A) levels using immunoblotting.
Results
Endothelial cells stimulated with insulin exhibited an increase in phosphorylation of Aktser 473 in serum free conditions but such insulin response was not observed in cells treated with IL-6, suggesting chronic exposure of endothelial cells to IL-6 leads to insulin resistance. HPLC analysis for global DNA methylation resulted in decreased levels of 5-methyl cytosine in cells treated with pro-inflammatory molecules (both by NCM and IL-6) as compared to untreated controls. Subsequently, analysis in cells treated with IL-6 showed a significant decrease in DNMT1 levels but not in DNMT3A. Other pro-inflammatory marker such as TNF-α did not exhibit such changes.
Conclusion
Our study suggests: a) Chronic treatment of endothelial cells with IL-6 results in insulin resistance b) Neutrophil conditioned medium and IL-6 decreases methyl cytosine levels c) DNMT1 but not DNMT3a levels are reduced in cells treated with IL-6.
Jaipal Panga Reddy, Dulal Panda and Sanjeeva Srivastava
The rapid emergence of microbial resistance against the ever increasing number of infectious diseases has been a major hurdle in hunt for novel antibiotics and inventive targets required to combat these dreaded pathogens. Despite the discovery of synthetic and semi-synthetic drugs, infectious diseases remain a major cause of concern. The process of drug discovery started from natural products as an antibacterial drug, an old art which was widely adopted in ancient civilizations in India and China. Most of the existing antibiotics are derived from the backbone of natural compounds (Butler M S 2005). Drug discovery process from natural products to synthetic medicine has continued for thousands of years to battle with pathogenic organisms. Although synthetic drugs played a vital role as antimicrobial drugs during the last decade, the over-use of antibiotics has led to the unanticipated changes at the genomic level, resulting into emergence of antibiotic-resistant strains (Singh P et al. 2010). To combat drug-resistant strains there is a need to develop and characterize new drugs by screening natural and synthetic compounds. Curcumin is a well known natural product, with a potential to cure wide range of cancers. Apart from antitumor activity, it also has anti-inflammatory, anti-viral, anti-genotoxic, phototoxic and antimicrobial activities. The exact mechanism of action of curcumin is still obscure and further investigations are required to identify its molecular/cellular targets. Recently, it was observed that curcumin is able to perturb the FtsZ assembly dynamics and elongate the bacterial cell length by inhibiting bacterial cell division (Rai D et al. 2008).
In the present study, we aimed at deciphering the mechanism of action and molecular targets of curcumin in B. subtilis AH75 using classical two dimensional electrophoresis, 2D-difference gel electrophoresis (2DDIGE) in combination with MALDI-TOF/TOFMS. Comparative proteome analysis of control and IC50 curcumin treated B. subtilis has revealed differential expression of 48 proteins (p ≤ 0.05) in response to curcumin treatment. Further, in silico analysis has revealed the involvement of the differential expressed proteins in protein synthesis, transcription/nucleotide synthesis, stress regulation, central metabolism and amino acid metabolic process. Additionally, suppression in major central metabolic dehydrogenase such as glyceraldehyde-3-phosphate dehydrogenase 2, dihydrolipoyl dehydrogenase, malate dehydrogenase, pyruvate dehydrogenase E1 component subunit beta, 2-oxoglutarate dehydrogenase E1 component, ATP synthase epsilon chain, ATP synthase subunit beta and probable NADdependent malic enzyme 1 has been identified (Figure 1). Reduction in expression levels of major dehydrogenases indicates the disturbance in cell membrane integrity to transfer the electrons from NADH to molecular oxygen or maintain the PMF or maintain the membrane potential. Selected potential proteins obtained from proteomic analysis were validated with real-time expression analysis and metabolic assays such as resazurin assay for metabolic activity, CTC assay for respiratory activity and propidium iodide staining for membrane integrity. Findings from this study have revealed that curcumin has potential effects on multiple physiological pathways in bacteria and inhibition of the cell division machinery is one of the prime targets of this drug. Multiple proteins involved in cell division process such as GroEL, ClpC, MetK, Tuf and major dehydrogenases are significantly modulated as a consequence of the drug treatment.