 Claudia AM Wheeler-Kingshott, Ph.D.
Claudia AM Wheeler-Kingshott, Ph.D.
University Reader in Magnetic Resonance Physics, Department of Neuroinflammation, UCL Institute of Neurology, London, UK
Abstract
Detecting neuronal activity in vivo non-invasively is possible with a number of techniques. Amongst these, in 1990 functional magnetic resonance imaging (fMRI) was proposed as a technique that has a great ability to spatially map brain activity by exploiting the blood oxygenation level dependent (BOLD) contrast mechanism [1, 2]. In fact, neuronal activation triggers a demand for oxygen and induces a localised increase in blood flow and blood volume, which actually exceeds the metabolic needs. This in turns causes an increase of oxyhaemoglobin in the venous compartment, which is a transient phenomenon and is accompanied by a transient change (decrease) in the concentration of deoxyhaemoglobin. Due to its paramagnetic properties, the amount of deoxyhaemoglobin present in the venous blood affects the local magnetic field seen by the spins (protons) and determines the local properties of the MR signal. A decrease in deoxyhaemoglobin during neuronal activity, therefore, induces local variations of this magnetic field that increases the average transverse relaxation time of tissue, measured via the T2* parameter [3]. This means that there is an increase of the MR signal (of the order of a few %, typically <5%) linked to metabolic changes happening during brain function. Activation can be inferred at different brain locations by performing tasks while acquiring the MR signal and comparing periods of rest to periods of activity.
The macroscopic changes of the BOLD signal are well characterised, while the reason for the increased blood supply, exceeding demands, needs further thoughts. Here we will discuss two approaches for explaining the BOLD phenomenon, one that links it to adenosine triphosphate production [4] and enzyme saturation, the other that relates it to the very slow diffusion of oxygen through the blood-brain-barrier with a consequent compensatory high demand of oxygen [5]. Some evidence of restricted oxygen diffusion has been shown by means of hypercapnia [6], although it is not excluded that both mechanisms may be present.
Overall, the BOLD signal changes theory and its physiological basis will be presented and discussed.
References
- Ogawa, S., et al., Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci U S A, 1990. 87(24): p. 9868-72.
- Kwong, K.K., et al., Dynamic magnetic resonance imaging of human brain activity during primary sensory stimulation. Proc Natl Acad Sci U S A, 1992. 89(12): p. 5675-9.
- Bandettini PA, et al. Spin-echo and gradient-echo EPI of human brain activation using BOLD contrast: a comparative study at 1.5 T. NMR Biomed. 1994 Mar;7(1-2):12-20
- Fox, P.T., et al., Nonoxidative glucose consumption during focal physiologic neural activity. Science, 1988. 241(4864): p. 462-4.
- Gjedde, A., et al. Reduction of functional capillary density in human brain after stroke. J Cereb Blood Flow Metab, 1990. 10(3): p. 317-26.
- Hoge, R.D., et al., Linear coupling between cerebral blood flow and oxygen consumption in activated human cortex. Proc Natl Acad Sci U S A, 1999. 96(16): p. 9403-8.
 Egidio D’Angelo, MD, Ph.D.
Egidio D’Angelo, MD, Ph.D.
Full Professor of Physiology & Director, Brain Connectivity Center, University of Pavia, Italy
Realistic modeling: new insight into the functions of the cerebellar network
Realistic modeling is an approach based on the careful reconstruction of neurons synapses starting from biological details at the molecular and cellular level. This technique, combined with the connection topologies derived from histological measurements, allows the reconstruction of precise neuronal networks. Finally, the advent of specific software platforms (PYTHON-NEURON) and of super-computers allows large-scale network simulation to be performed in reasonable time. This approach inverts the logics of older theoretical models, which anticipated an intuition on how the network might work. In realistic modeling, network properties “emerge” from the numerous biological properties embedded into the model.
This approach is illustrated here through an outstanding application of realistic modeling to the cerebellar cortex network. The neurons (over 105) are reproduced at a high level of detail generating non-linear network effects like population oscillations and resonance, phase-reset, bursting, rebounds, short-term and long-term plasticity, spatiotemporal redistrbution of input patterns. The model is currently being used in the context of he HUMAN BRAIN PROJECT to investigate the cerebellar network function.
Correspondence should be addressed to
Dr. EgidioD’Angelo,
Laboratory of Neurophysiology
Via Forlanini 6, 27100 Pavia, Italy
Phone: 0039 (0) 382 987606
Fax: 0039 (0) 382 987527
dangelo@unipv.it
Acknowledgments
This work was supported by grants from European Union to ED (CEREBNET FP7-ITN238686, REALNET FP7-ICT270434) and by grants from the Italian Ministry of Health to ED (RF-2009-1475845).

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
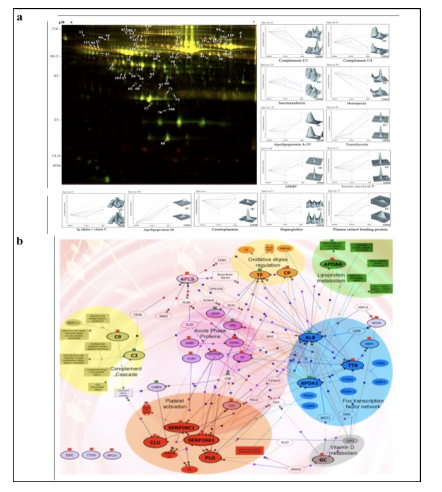
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Kal Ramnarayan, Ph.D.
Kal Ramnarayan, Ph.D.
Co-founder President & Chief Scientific Officer, Sapient Discovery, San Diego, CA, USA
A cost-effective approach to Protein Structure-guided Drug Discovery: Aided by Bioinformatics, Chemoinformatics and computational chemistry
With the mapping of the human genome completed almost a decade ago, efforts are still underway to understand the gene products (i.e., proteins) in the human biological and disease pathways. Deciphering such information is very important for the discovery and development of small molecule drugs as well as protein therapeutics for various human diseases for which no cure exists. As an example, with more than 500 members, the kinase family of protein targets continues to be an important and attractive class for drug discovery. While how many of the members in this family are actually druggable is still to be established, there are several ongoing efforts on this class of proteins across a broad spectrum of disease categories. Even though in general the protein structural topology might looks similar, there are issues with respect selectivity of identified small molecule inhibitors when, the lead molecule discovery is carried out at the ATP binding site. As an added complexity, allosteric modulators are needed for some of the members, but the actual site for such modulation on the protein target can not resolved with uncertainty. In this presentation we will describe a bioinformatics and computational based platform for small molecule discovery for protein targets that are involved in protein-protein interactions as well as targets like kinases and phosphatases. We will describe a computational approach in which we have used an informatics based platform with several hundred kinases to sort through in silico and identify inhibitors that are likely to be highly selective in the lead generation phase. We will discuss the implication of this approach on the drug discovery of the kinase and phosphatase classes in general and independent of the disease category.

Arathy R and Binoy B Nair
PC based heart sound monitoring system
Heart diseases caused by disorders of the heart and blood vessels, are world’s largest killers. Early detection and monitoring of heart abnormalities is essential for diagnosis and effective treatment of heart diseases. Severalmethodologies are used for screening and diagnosing heart diseases. They are auscultation, electrocardiogram (ECG), echo-cardiogram, ultrasound etc. The effectiveness and applicability of all these diagnostic methods are highly dependent on the equipment cost and size as well as skill of the physician. This paper presents the design and development of a low cost portable wireless/tubeless digital stethoscope which can be used by the physician for monitoring the patient from a distance. The stethoscope system interfaces to a PC and is also capable of analyzing the heart sounds and identifying abnormalities in the heart sound and its classification. Storage of heart sound for later analysis is also possible.This advanced functionality increases the physician’s diagnostic capability, and such a PCG is not still available in most hospitals. Acoustic stethoscope can be changed into a digital stethoscope by inserting an electric capacity microphone into its diaphragm (Wang, Chen and Samjin, 2009).
![Delegate Talk: Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors @ Sathyam Hall | Vallikavu | Kerala | India](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/bicb.jpg)
Rajasekhar Chekkara, Venkata Reddy Gorla and Sobha Rani Tenkayala
Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors
Polo-like kinase 1 (PLK1) is a significant enzyme with diverse biological actions in cell cycle progression, specifically mitosis. Suppression of PLK1 activity by small molecule inhibitors has been shown to inhibit cancer, being BI 2536 one of the most potent active inhibitor of PLK1 mechanism. Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies were carried out for a set of 54 compounds belonging to Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK1 inhibitors. A six-point pharmacophoremodel AAADDR, with three hydrogen bond acceptors (A), two hydrogen bond donors (D) and one aromatic ring (R) was developed by Phase module of Schrdinger suite Maestro 9. The generated pharmacophore model was used to derive a predictive atom-based 3D quantitative structure-activity relationship analysis (3D-QSAR) model for the training set (r2 = 0.88, SD = 0.21, F = 57.7, N = 44) and for test set (Q2 = 0.51, RMSE = 0.41, PearsonR = 0.79, N = 10). The original set of compounds were docked into the binding site of PLK1 using Glide and the active residues of the binding site were analyzed. The most active compound H18 interacted with active residues Leu 59, Cys133 (glide score = −10.07) and in comparison of BI 2536, which interacted with active residues Leu 59, Cys133 (glide score = −10.02). The 3D-QSAR model suggests that hydrophobic and electron-withdrawing groups are essential for PLK1 inhibitory activity. The docking results describes the hydrogen bond interactions with active residues of these compounds. These results which may support in the design and development of novel PLK1 inhibitors.