





















 Aditya Murthy, Ph.D.
Aditya Murthy, Ph.D.
Associate Professor, Centre For Neuroscience, Indian Institute of Science, Bangalore, India
Since Karl Lashley’s seminal work on the formulation of serial order, numerous models assume simultaneous representation of competitive elements of a sequence, to account for serial order effects in different types of behavior like typing, speech, etc. Such models follow two basic assumptions: (1) more than one plan representation can be simultaneously active in a planning layer; (2) the most active plan is chosen in another layer called the competitive choice layer. Using the oculomotor system I will describe behavioral and neurophysiological experiments that tests the two critical predictions of such queuing models, providing evidence that basal ganglia in monkeys and humans instantiate a form of queuing that transforms parallel movement representations into more serial representations, allowing for the expression of sequential saccadic eye movements.

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
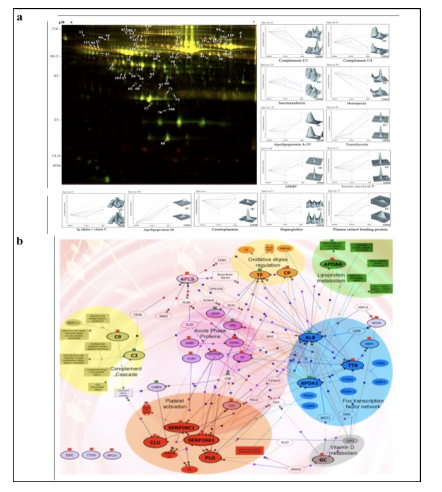
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Kal Ramnarayan, Ph.D.
Kal Ramnarayan, Ph.D.
Co-founder President & Chief Scientific Officer, Sapient Discovery, San Diego, CA, USA
A cost-effective approach to Protein Structure-guided Drug Discovery: Aided by Bioinformatics, Chemoinformatics and computational chemistry
With the mapping of the human genome completed almost a decade ago, efforts are still underway to understand the gene products (i.e., proteins) in the human biological and disease pathways. Deciphering such information is very important for the discovery and development of small molecule drugs as well as protein therapeutics for various human diseases for which no cure exists. As an example, with more than 500 members, the kinase family of protein targets continues to be an important and attractive class for drug discovery. While how many of the members in this family are actually druggable is still to be established, there are several ongoing efforts on this class of proteins across a broad spectrum of disease categories. Even though in general the protein structural topology might looks similar, there are issues with respect selectivity of identified small molecule inhibitors when, the lead molecule discovery is carried out at the ATP binding site. As an added complexity, allosteric modulators are needed for some of the members, but the actual site for such modulation on the protein target can not resolved with uncertainty. In this presentation we will describe a bioinformatics and computational based platform for small molecule discovery for protein targets that are involved in protein-protein interactions as well as targets like kinases and phosphatases. We will describe a computational approach in which we have used an informatics based platform with several hundred kinases to sort through in silico and identify inhibitors that are likely to be highly selective in the lead generation phase. We will discuss the implication of this approach on the drug discovery of the kinase and phosphatase classes in general and independent of the disease category.
 Karmeshu, Ph.D.
Karmeshu, Ph.D.
Dean & Professor, School of Computer & Systems Sciences & School of Computational & Integrative Sciences, Jawaharlal Nehru University, India.
Interspike Interval Distribution of Neuronal Model with distributed delay: Emergence of unimodal, bimodal and Power law
The study of interspike interval distribution of spiking neurons is a key issue in the field of computational neuroscience. A wide range of spiking patterns display unimodal, bimodal ISI patterns including power law behavior. A challenging problem is to understand the biophysical mechanism which can generate the empirically observed patterns. A neuronal model with distributed delay (NMDD) is proposed and is formulated as an integro-stochastic differential equation which corresponds to a non-markovian process. The widely studied IF and LIF models become special cases of this model. The NMDD brings out some interesting features when excitatory rates are close to inhibitory rates rendering the drift close to zero. It is interesting that NMDD model with gamma type memory kernel can also account for bimodal ISI pattern. The mean delay of the memory kernels plays a significant role in bringing out the transition from unimodal to bimodal ISI distribution. It is interesting to note that when a collection of neurons group together and fire together, the ISI distribution exhibits power law.
 Akhilesh Pandey, Ph.D.
Akhilesh Pandey, Ph.D.
Professor, Johns Hopkins University School of Medicine, Baltimore, USA
A draft map of the human proteome
We have generated a draft map of the human proteome through a systematic and comprehensive analysis of normal human adult tissues, fetal tissues and hematopoietic cells as an India-US initiative. This unique dataset was generated from 30 histologically normal adult tissues, fetal tissues and purified primary hematopoietic cells that were analyzed at high resolution in the MS mode and by HCD fragmentation in the MS/MS mode on LTQ-Orbitrap Velos/Elite mass spectrometers. This dataset was searched against a 6-frame translation of the human genome and RNA-Seq transcripts in addition to standard protein databases. In addition to confirming a large majority (>16,000) of the annotated protein-coding genes in humans, we obtained novel information at multiple levels: novel protein-coding genes, unannotated exons, novel splice sites, proof of translation of pseudogenes (i.e. genes incorrectly annotated as pseudogenes), fused genes, SNPs encoded in proteins and novel N-termini to name a few. Many proteins identified in this study were identified by proteomic methods for the first time (e.g. hypothetical proteins or proteins annotated based solely on their chromosomal location). We have generated a catalog of proteins that show a more tissue-restricted pattern of expression, which should serve as the basis for pursuing biomarkers for diseases pertaining to specific organs. This study also provides one of the largest sets of proteotypic peptides for use in developing MRM assays for human proteins. Identification of several novel protein-coding regions in the human genome underscores the importance of systematic characterization of the human proteome and accurate annotation of protein-coding genes. This comprehensive dataset will complement other global HUPO initiatives using antibody-based as well as MRM mass spectrometry-based strategies. Finally, we believe that this dataset will become a reference set for use as a spectral library as well as for interesting interrogations pertaining to biomedical as well as bioinformatics questions.


Tejaswini Subbannayya, Nandini A. Sahasrabuddhe, Arivusudar Marimuthu, Santosh Renuse, Gajanan Sathe, Srinivas M. Srikanth, Mustafa A. Barbhuiya, Bipin Nair, Juan Carlos Roa, Rafael Guerrero-Preston, H. C. Harsha, David Sidransky, Akhilesh Pandey, T. S. Keshava Prasad and Aditi Chatterjee
Proteomic profiling of gallbladder cancer secretome – a source for circulatory biomarker discovery
Gallbladder cancer (GBC) is the fifth most common cancer of the gastrointestinal tract and one of the common malignancies that occur in the biliary tract (Misra et al. 2006; Lazcano-Ponce et al. 2001). It has a poor prognosis with survival of less than 5 years in 90% of the cases (Misra et al. 2003). The etiology is ill-defined. Several risk factors have been reported including cholelithiasis, obesity, female gender and exposure to carcinogens (Eslick 2010; Kumar et al. 2006). Poor prognosis in GBC is mainly due to late presentation of the disease and lack of reliable biomarkers for early diagnosis. This emphasizes the need to identify and characterize cancer biomarkers to aid in the diagnosis and prognosis of GBC. Secreted proteins are an important class of molecules which can be detected in body fluids and has been targeted for biomarker discovery. There are challenges faced in the proteomic interrogation of body fluids especially plasma such as low abundance of tumor secreted proteins, high complexity and high abundance of other proteins that are not released by the tumor cells (Tonack et al. 2009). Profiling of conditioned media from the cancer cell lines can be used as an alternate means to identify secreted proteins from tumor cells (Kashyap et al. 2010; Marimuthu et al. 2012). We analyzed the invasive property of 7 GBC cell lines (SNU-308, G-415, GB-d1, TGBC2TKB, TGBC24TKB, OCUG-1 and NOZ). Four cell lines were selected for analysis of the cancer secretome based on the invasive property of the cells. We employed isobaric tags for relative and absolute quantitation (iTRAQ) labeling technology coupled with high resolution mass spectrometry to identify and characterize secretome from the panel of 4GBC cancer cells mentioned above. In total, we have identified around 2,000 proteins of which 175 were secreted at differential abundance across all the four cell lines. This secretome analysis will act as a reservoir of candidate biomarkers. Currently, we are investigating and validating these candidate markers from GBC cell secretome. Through this study, we have shown mass spectrometry-based quantitative proteomic analysis as a robust approach to investigate secreted proteins in cancer cells.