 Upinder S. Bhalla, Ph.D.
Upinder S. Bhalla, Ph.D.
Professor & Dean, NCBS, Bengaluru, India
Watching the network change during the formation of associative memory
The process of learning is measured through behavioural changes, but it is of enormous interest to understand its cellular and network basis. We used 2-photon imaging of hippocampal CA1 pyramidal neuron activity in mice to monitor such changes during the acquisition of a trace conditioning task. One of the questions in such learning is how the network retains a trace of a brief conditioned stimulus (a sound), until the arrival of a delayed unconditioned stimulus (a puff of air to the eye). During learning, the mice learn to blink when the tone is presented, well before the arrival of the air puff.
The mice learnt this task in 20-50 trials. We observed that in this time-frame the cells in the network changed the time of their peak activity, such that their firing times tiled the interval between sound and air puff. Thus the cells seem to form a relay of activity. We also observed an evolution in functional connectivity in the network, as measured by groupings of correlated cells. These groupings were stable till the learning protocol commenced, and then changed. Thus we have been able to observe two aspects of network learning: changes in activity (relay firing), and changes in connectivity (correlation groups).


 Ashok Pandey, Ph.D.
Ashok Pandey, Ph.D.
Scientist F & Head, Biotechnology Division, National Institute for Interdisciplinary Science and Technology-CSIR), Thiruvananthapuram, India
Alternative renewable resources: Issues and perspectives for India – the case of transport fuels
With the increase in the urbanization way of life and also more and more dependence on materialistic life, there is substantial growing demand for the energy. The science and technological policy of the India has looked several avenues to fulfill this demand through alternative resources such as solar energy, wind energy, tidal energy, bioenergy, etc. The demand for the transport sector is largely met through the import (~70%). Biofuels, in particular bioethanol from lignocellulosic biomass offer attractive possibilities in this regard.
The sugar platform which generates ethanol is considered to be the most valuable solution to the transport fuel demand. Bioethanol can be generated from grains as well as from lignocellulosic plant material by their saccharification to sugars and subsequent fermentation of the sugars to produce ethanol. Bio-ethanol as a transportation fuel is attractive since it is more energy efficient than gasoline and produces less emissions. The benefits of developing biomass to ethanol technology(s) include: increased national energy security, reduction in GHG emissions, use of renewable resources, economic benefits and creation of employment and the foundation of a carbohydrate based chemical industry. However, the utilization of lignocellulosic biomass for fuel generation has not been given the sort of attention it ought to receive. It is known that the technology for ethanol production from biomass has to evolve greatly for an economical commercial scale utilization of the renewable biomass resources. Biomass requires extensive processing involving multiple steps for hydrolysis and fermentation of the raw material for producing ethanol. Feed stock availability, pretreatment, saccharification, fermentation and ethanol recovery are all factors which influence the production of ethanol and which needs R&D efforts for overall improvement of the production economics.
Bioconversion of lignocellulosic biomass (LB) can contribute significantly to the production of organic chemicals also. LB is also considered to be the only foreseeable source of energy. LB is mainly composed of (dry wt basis): cellulose, 40-60; hemicellulose, 20-40; and lignin, 10-25%. Most efficient method of biomass hydrolysis is through enzymatic saccharification5 using cellulases and hemicellulases. Fungal cellulases (FCs) have proved to be a better candidate than other microbial cellulases, with their secreted free cellulase complexes comprising all three components of cellulase [endoglucanases, exoglucanases and cellobiases (glucosidases).
The Centre for Biofuels at NIIST, Trivandrum, India aims ultimately to develop technologies and processes which will address the nation’s need for making fuel ethanol from the renewable resource: biomass. It is proposed to direct R&D activities at the major requirements of a biomass-ethanol technology, which include production of cellulases, hydrolysis of biomass, and ethanol fermentation. Viable technologies for each of these processes will contribute to the overall process development for fuel alcohol production from cheap and renewable biomass resources.
The lecture would present perspectives on bioethanol from lignocellulosic feedstocks.
References
- Biofuels- Alternative Feedstocks and Conversion Processes, Editors- Ashok Pandey, C Larroche, SC Ricke, CG Dussap & E Gnansounou, Academic Press, Elsevier Inc; San Diego, USA, p629 (2011) ISBN: 978-0-12-385099-7
- Handbook of Plant-Based Biofuels, Editor- Ashok Pandey, CRC Press, Francis & Taylors, Boca Raton, USA, p 297 (2008) ISBN 978-q-5602-2175-3
- Biofuels II, Special issue of Journal of Scientific & Industrial Research, Guest Editors- E Gnansounou, C Larroche and Ashok Pandey, 67(11), 837-1040 (2008) ISSN: 0022-4456
- Biofuels, Special issue of Journal of Scientific & Industrial Research, Guest Editors- C Larroche and Ashok Pandey, 64(11), 797-988 (2005) ISSN: 0022-4456

 K. Satyamoorthy, Ph.D.
K. Satyamoorthy, Ph.D.
Director, Life Sciences Centre, Manipal University, India
Epigenetic Changes due to DNA Methylation in Human Epithelial Tumors
Extensive global hypomethylation in the genome and hypermthylation of selective tumor specific suppressor genes appears to be a hallmark of human cancers. Data suggests that hypermethylation of promoter region in genes is more closely related to subsequent gene expression; contrary to gene-body DNA methylation. The intricate balance between these two may contribute to the progressive process of development, differentiation and carcinogenesis. Epigenetic changes encompass, apart from DNA methylation, chromatin modifications through post-translational changes in histones and control by miRNAs. At the genome level, effects from these are compounded by copy number variations (CNVs) which may ultimately influence protein functions. From clinical perspective, changes in DNA methylation occur very early which are reversible and are influenced by environmental factors. Therefore, these can be potential resource for identifying therapeutic targets as well as biomarkers for early screening of cancer. Our current efforts in profiling genome wide DNA methylation changes in oral, cervical and breast cancers through DNA methylation microarray analysis has revealed number of alterations critical for survival, progression and metastatic behavior of tumors. Bioinformatics and functional analysis revealed several key regulatory molecules controlled by DNA methylation and suggests that DNA methylation changes in several CpG islands appear to co-segregate in the regions of miRNAs as well as in the CNVs. We have validated the signatures for methylation of CpG islands through bisufite sequencing for essential genes in clinical samples and have undertaken transcriptional and functional analysis in tumor cell lines. These results will be presented.
 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
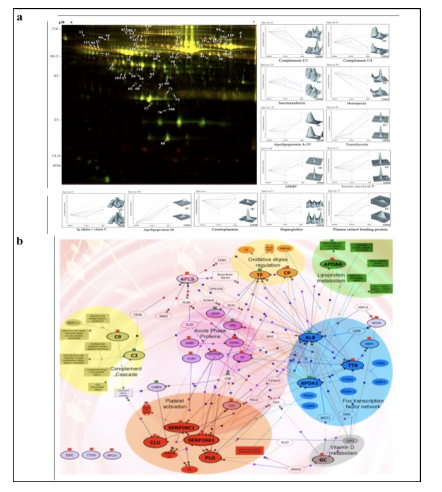
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Andrey Panteleyev, Ph.D.
Andrey Panteleyev, Ph.D.
Vice Chair, Division of Molecular Biology, NBICS Centre-Kurchatov Institute, Moscow, Russia
The system of PAS proteins (HIF and AhR) as an interface between environment and skin homeostasis
Regulation of normal skin functions as well as etiology of many skin diseases are both tightly linked to the environmental impact. Nevertheless, molecular aspects of skin-environment communication and mechanisms coordinating skin response to a plurality of environmental stressors remain poorly understood.
Our studies along with the work of other groups have identified the family of PAS dimeric transcription factors as an essential sensory and regulatory component of communication between skin and the environment. This protein family comprises a number of hypoxia-induced factors (HIF-alpha proteins), aryl hydrocarbon receptor (AhR), AhR nuclear translocator (ARNT), and several proteins implicated in control of rhythmic processes (Clock, Period, and Bmal proteins). Together, various PAS proteins (and first of all ARNT – as the central dimerization partner in the family) control such pivotal aspects of cell physiology as drug/xenobiotic metabolism, hypoxic and UV light response, ROS activity, pathogen defense, overall energy balance and breathing pathways.
In his presentation Dr. Panteleyev will focus on the role of ARNT activity and local hypoxia in control of keratinocyte differentiation and cornification. His recent work revealed that ARNT negatively regulates expression of late differentiation genes through modulation of amphiregulin expression and downstream alterations in activity of EGFR pathway. All these effects are highly dependent on epigenetic mechanisms such as histone deacetylation. Characterisation of hypoxia as a key microenvironmental factor in the skin and the role of HIF pathway in control of dermal vasculature and epidermal functions is another major focus of Dr. Panteleyev’s presentation.
In general, the studies of Dr. Panteleyev’s laboratory provide an insight into the PAS-dependent maintenance of skin homeostasis and point to the potential role of these proteins in pathogenesis of environmentally-modulated skin diseases such as barrier defects, desquamation abnormalities, psoriasis, etc.
 Sudarslal S, Ph.D.
Sudarslal S, Ph.D.
Associate Professor, School of Biotechnology, Amrita University
Electrospray ionization ion trap mass spectrometry for cyclic peptide characterization
There has been considerable interest in the isolation and structural characterization of bioactive peptides produced by bacteria and fungi. Most of the peptides are cyclic depsipeptides characterized by the presence of lactone linkages and β-hydroxy fatty acids. Occurrence of microheterogeneity is another remarkable property of these peptides. Even if tandem mass spectrometers are good analytical tools to structurally characterize peptides and proteins, sequence analysis of cyclic peptides is often ambiguous due to the random ring opening of the peptides and subsequent generation of a set of linear precursor ions with the same m/z. Here we report combined use of chemical derivatization and multistage fragmentation capability of ion trap mass spectrometers to determine primary sequences of a series of closely related cyclic peptides.



Ravindra Gudihal, Suresh Babu C V
Bioanalytical Characterization of Therapeutic Proteins
The characterization of therapeutic proteins such as monoclonal antibody (mAb) during different stages of manufacturing is crucial for timely and successful product release. Regulatory agencies require a variety of analytical technologies for comprehensive and efficient protein analysis. Electrophoresis-based techniques and liquid chromatography (LC) either standalone or coupled to mass spectrometry (MS) are at the forefront for the in-depth analysis of protein purity, isoforms, stability, aggregation, posttranslational modifications, PEGylation, etc. In this presentation, a combination of various chromatographic and electrophoretic techniques such as liquid-phase isoelectric focusing, microfluidic and capillary-based electrophoresis (CE), liquid chromatography (LC) and combinations of those with mass spectrometry techniques will be discussed. We present a workflow based approach to the analysis of therapeutic proteins. In successive steps critical parameters like purity, accurate mass, aggregation, peptide sequence, glycopeptide and glycan analysis are analyzed. In brief, the workflow involved proteolytic digestion of therapeutic protein for peptide mapping, N-Glycanase and chemical labeling reaction for glycan analysis, liquid-phase isoelectric focusing for enrichment of charge variants followed by a very detailed analysis using state of the art methods such as CE-MS and LC-MS. For the analysis of glycans, we use combinations of CE-MS and LC-MS to highlight the sweet spots of these techniques. CE-MS is found to be more useful in analysis of highly sialylated glycans (charged glycans) while nano LC-MS seems to be better adapted for analysis of neutral glycans. These two techniques can be used to get complementary data to profile all the glycans present in a given protein. In addition, microfluidic electrophoresis was used as a QC tool in initial screening for product purity, analysis of papain digestion fragments of mAb, protein PEGylation products, etc. The described workflow involves multiple platforms, provides an end to end solution for comprehensive protein characterization and aims at reducing the total product development time.