 Gaute Einevoll, Ph.D.
Gaute Einevoll, Ph.D.
Professor of Physics, Department of Mathematical Sciences & Technology, Norwegian University of Life Sciences (UMB)
Multiscale modeling of cortical network activity: Key challenges
Gaute T. Einevoll Computational Neuroscience Group, Norwegian University of Life Sciences, 1432 Ås, Norway; Norwegian National Node of the International Neuroinformatics Coordinating Facility (INCF)
Several challenges must be met in the development of multiscale models of cortical network activity. In the presentation I will, based on ongoing work in our group (http://compneuro.umb.no/ ) on multiscale modeling of cortical columns, outline some of them. In particular,
-
Combined modeling schemes for neuronal, glial and vascular dynamics [1,2],
-
Development of sets of interconnected models describing system at different levels of biophysical detail [3-5],
-
Multimodal modeling, i.e., how to model what you can measure [6-12],
-
How to model when you don’t know all the parameters, and
-
Development of neuroinformatics tools and routines to do simulations efficiently and accurately [13,14].
References:
- L. Øyehaug, I. Østby, C. Lloyd, S.W. Omholt, and G.T. Einevoll: Dependence of spontaneous neuronal firing and depolarisation block on astroglial membrane transport mechanisms, J Comput Neurosci 32, 147-165 (2012)
- I. Østby, L. Øyehaug, G.T. Einevoll, E. Nagelhus, E. Plahte, T. Zeuthen, C. Lloyd, O.P. Ottersen, and S.W. Omholt: Astrocytic mechanisms explaining neural-activity-induced shrinkage of extraneuronal space, PLoS Comp Biol 5, e1000272 (2009)
- T. Heiberg, B. Kriener, T. Tetzlaff, A. Casti, G.T. Einevoll, and H.E. Plesser: Firing-rate models can describe the dynamics of the retina-LGN connection, J Comput Neurosci (2013)
- T. Tetzlaff, M. Helias, G.T. Einevoll, and M. Diesmann: Decorrelation of neural-network activity by inhibitory feedback, PLoS Comp Biol 8, e10002596 (2012).
- E. Nordlie, T. Tetzlaff, and G.T. Einevoll: Rate dynamics of leaky integrate-and-fire neurons with strong synapses, Frontiers in Comput Neurosci 4, 149 (2010)
- G.T. Einevoll, F. Franke, E. Hagen, C. Pouzat, K.D. Harris: Towards reliable spike-train recording from thousands of neurons with multielectrodes, Current Opinion in Neurobiology 22, 11-17 (2012)
- H. Linden, T Tetzlaff, TC Potjans, KH Pettersen, S Grun, M Diesmann, GT Einevoll: Modeling the spatial reach of LFP, Neuron 72, 859-872 (2011).
- H. Linden, K.H. Pettersen, and G.T. Einevoll: Intrinsic dendritic filtering gives low-pass power spectra of local field potentials, J Computational Neurosci 29, 423-444 (2010)
- K.H. Pettersen and G.T. Einevoll: Amplitude variability and extracellular low-pass filtering of neuronal spikes, Biophysical Journal 94, 784-802 (2008).
- K.H. Pettersen, E. Hagen, and G.T. Einevoll: Estimation of population firing rates and current source densities from laminar electrode recordings, J Comput Neurosci 24, 291-313 (2008).
- K. Pettersen, A. Devor, I. Ulbert, A.M. Dale and G.T. Einevoll. Current-source density estimation based on inversion of electrostatic forward solution: Effects of finite extent of neuronal activity and conductivity discontinuities, Journal of Neuroscience Methods 154, 116-133 (2006).
- G.T. Einevoll, K. Pettersen, A. Devor, I. Ulbert, E. Halgren and A.M. Dale: Laminar Population Analysis: Estimating firing rates and evoked synaptic activity from multielectrode recordings in rat barrel cortex, Journal of Neurophysiology 97, 2174-2190 (2007).
- LFPy: A tool for simulation of extracellular potentials (http://compneuro.umb.no)
- E. Nordlie, M.-O. Gewaltig, H. E. Plesser: Towards reproducible descriptions of neuronal network models, PLoS Comp Biol 5, e1000456 (2009).

 Bharat B. Chattoo, Ph.D.
Bharat B. Chattoo, Ph.D.
Professor, Faculty of Science M.S.University of Baroda, India
Biology of plant infection by Magnaporthe oryzae
The rice blast disease caused by the ascomycetous fungus Magnaporthe oryzae is a major constraint in rice production. Rice-M.oryzae is also emerging as a good model patho-system to investigate how the fungus invades and propagates within the host. Identification and characterisation of genes critical for fungal pathogenesis provides opportunities to explore their use as possible targets for development of strategies for combating fungal infection and to better understand the complex process of host-pathogen interaction.
We have used insertional mutagenesis and RNAi based approaches to identify pathogenesis related genes in this fungus. A large number of mutants were isolated using Agrobacterium tumefaciens mediated transformation (ATMT). Characterisation of several interesting mutants is in progress. We have identified a novel gene, MGA1, required for the development of appressoria. The mutant mga1 is unable to infect and is impaired in glycogen and lipid mobilization required for appressorium development. The glycerol content in the mycelia of the mutant was significantly lower as compared to wild type and it was unable to tolerate hyperosmotic stress. A novel ABC transporter was identified in this fungus. The abc4 mutant did not form functional appressoria, was non-pathogenic and showed increased sensitivity to certain antifungal molecules implying the role of ABC4 in multidrug resistance (MDR). Another mutant MoSUMO (MGG_05737) was isolated using a Split Marker technique; the mutant showed defects in growth, germination and infection. Immuno-fluorescence microscopy revealed that MoSumo is localized to septa in mycelia and nucleus as well as septa in spores. Two Dimensional Gel Electrophoresis showed differences in patterns of protein expression between Wild Type B157 and MoΔSumo mutant. We also isolated and charaterised mutants in MoALR2 (MGG_08843) and MoMNR2 (MGG_09884). Our results indicate that both MoALR2 and MoMNR2 are Mg2+ transporters, and the reduction in the levels of CorA transporters caused defects in surface hydrophobicity, cell wall stress tolerance, sporulation, appressorium formation and infection are mediated through changes in the key signaling cascades in the knock-down transformants. (Work supported by the Department of Biotechnology, Government of India)
 Gillian Murphy, Ph.D.
Gillian Murphy, Ph.D.
Professor, Department of Oncology, University of Cambridge, UK
A novel strategy for targeting metalloproteinases in cancer
Epithelial tumours evolve in a multi-step manner, involving both inflammatory and mesenchymal cells. Although intrinsic factors drive malignant progression, the influence of the micro-environment of neoplastic cells is a major feature of tumorigenesis. Extracellular proteinases, notably the metalloproteinases, are key players in the regulation of this cellular environment, acting as major effectors of both cell-cell and cell-extracellular matrix (ECM) interactions. They are involved in modifying ECM integrity, growth factor availability and the function of cell surface signalling systems, with consequent effects on cellular differentiation, proliferation and apoptosis.This has made metalloproteinases important targets for therapeutic interventions in cancer and small molecule inhibitors focussed on chelation of the active site zinc and binding within the immediate active site pocket were developed. These were not successful in early clinical trials due to the relative lack of specificity and precise knowledge of the target proteinase(s) in specific cancers. We can now appreciate that it is essential that we understand the relative roles of the different enzymes (of which there are over 60) in terms of their pro and anti tumour activity and their precise sites of expression The next generations of metalloproteinase inhibitors need the added specificity that might be gained from an understanding of the structure of individual active sites and the role of extra catalytic domains in substrate binding and other aspects of their biology. We have prepared scFv antibodies to the extra catalytic domains of two membrane metalloproteinases, MMP-14 and ADAM17, that play key roles in the tumour microenvironment. Our rationale and experiences with these agents will be presented in more detail.

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
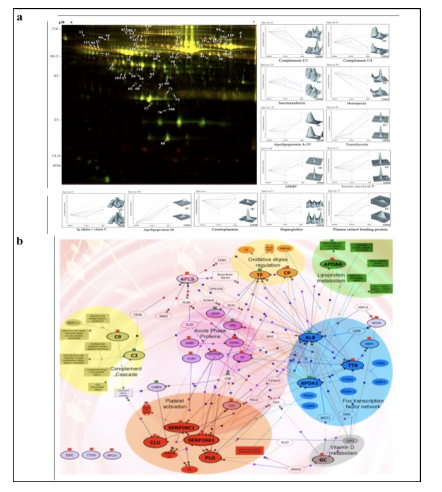
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)



David Ibanez, Laura Dubreuil and Alejandro Rier
Neurofeedback (NF) is a type of biofeedback that uses real time display of electroencephalography to illustrate brain activity. EEG features are extracted and displayed allowing the user to, with practice, modulate their temporal evolution. Neurofeedback training has many therapeutic applications such as attention deficit hyperactivity disorder (ADHD), migraine, depression or conduct disorders. This document presents NeuroSurfer, a novel general-purpose tool for neurofeedback training with a use case of attention deficit hyperactivity disorder (ADHD) treatment.
 Jaap Heringa, Ph.D.
Jaap Heringa, Ph.D.
Director & Professor of Bioinformatics, IBIVU VU University Amsterdam, The Netherlands
Modeling strategy based on Petri-nets
In my talk I will introduce a formal modeling strategy based on Petri-nets, which are a convenient means of modeling biological processes. I will illustrate the capabilities of Petri-nets as reasoning vehicles using two examples: Haematopoietic stem cell differentiation in mice, and vulval development in C. elegance. The first system was modeled using a Boolean implementation, and the second using a coarse-grained multi-cellular Petri-net model. Concepts such as the model state space, attractor states, and reasoning to adapt the model to the biological reality will be discussed.

 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
 Akhilesh Pandey, Ph.D.
Akhilesh Pandey, Ph.D.
Professor, Johns Hopkins University School of Medicine, Baltimore, USA
A draft map of the human proteome
We have generated a draft map of the human proteome through a systematic and comprehensive analysis of normal human adult tissues, fetal tissues and hematopoietic cells as an India-US initiative. This unique dataset was generated from 30 histologically normal adult tissues, fetal tissues and purified primary hematopoietic cells that were analyzed at high resolution in the MS mode and by HCD fragmentation in the MS/MS mode on LTQ-Orbitrap Velos/Elite mass spectrometers. This dataset was searched against a 6-frame translation of the human genome and RNA-Seq transcripts in addition to standard protein databases. In addition to confirming a large majority (>16,000) of the annotated protein-coding genes in humans, we obtained novel information at multiple levels: novel protein-coding genes, unannotated exons, novel splice sites, proof of translation of pseudogenes (i.e. genes incorrectly annotated as pseudogenes), fused genes, SNPs encoded in proteins and novel N-termini to name a few. Many proteins identified in this study were identified by proteomic methods for the first time (e.g. hypothetical proteins or proteins annotated based solely on their chromosomal location). We have generated a catalog of proteins that show a more tissue-restricted pattern of expression, which should serve as the basis for pursuing biomarkers for diseases pertaining to specific organs. This study also provides one of the largest sets of proteotypic peptides for use in developing MRM assays for human proteins. Identification of several novel protein-coding regions in the human genome underscores the importance of systematic characterization of the human proteome and accurate annotation of protein-coding genes. This comprehensive dataset will complement other global HUPO initiatives using antibody-based as well as MRM mass spectrometry-based strategies. Finally, we believe that this dataset will become a reference set for use as a spectral library as well as for interesting interrogations pertaining to biomedical as well as bioinformatics questions.
