 Bharat B. Chattoo, Ph.D.
Bharat B. Chattoo, Ph.D.
Professor, Faculty of Science M.S.University of Baroda, India
Biology of plant infection by Magnaporthe oryzae
The rice blast disease caused by the ascomycetous fungus Magnaporthe oryzae is a major constraint in rice production. Rice-M.oryzae is also emerging as a good model patho-system to investigate how the fungus invades and propagates within the host. Identification and characterisation of genes critical for fungal pathogenesis provides opportunities to explore their use as possible targets for development of strategies for combating fungal infection and to better understand the complex process of host-pathogen interaction.
We have used insertional mutagenesis and RNAi based approaches to identify pathogenesis related genes in this fungus. A large number of mutants were isolated using Agrobacterium tumefaciens mediated transformation (ATMT). Characterisation of several interesting mutants is in progress. We have identified a novel gene, MGA1, required for the development of appressoria. The mutant mga1 is unable to infect and is impaired in glycogen and lipid mobilization required for appressorium development. The glycerol content in the mycelia of the mutant was significantly lower as compared to wild type and it was unable to tolerate hyperosmotic stress. A novel ABC transporter was identified in this fungus. The abc4 mutant did not form functional appressoria, was non-pathogenic and showed increased sensitivity to certain antifungal molecules implying the role of ABC4 in multidrug resistance (MDR). Another mutant MoSUMO (MGG_05737) was isolated using a Split Marker technique; the mutant showed defects in growth, germination and infection. Immuno-fluorescence microscopy revealed that MoSumo is localized to septa in mycelia and nucleus as well as septa in spores. Two Dimensional Gel Electrophoresis showed differences in patterns of protein expression between Wild Type B157 and MoΔSumo mutant. We also isolated and charaterised mutants in MoALR2 (MGG_08843) and MoMNR2 (MGG_09884). Our results indicate that both MoALR2 and MoMNR2 are Mg2+ transporters, and the reduction in the levels of CorA transporters caused defects in surface hydrophobicity, cell wall stress tolerance, sporulation, appressorium formation and infection are mediated through changes in the key signaling cascades in the knock-down transformants. (Work supported by the Department of Biotechnology, Government of India)
 Rohit Manchanda, Ph.D.
Rohit Manchanda, Ph.D.
Professor, Biomedical Engineering Group, IIT-Bombay, India
Modelling the syncytial organization and neural control of smooth muscle: insights into autonomic physiology and pharmacology
We have been studying computationally the syncytial organization and neural control of smooth muscle in order to help explain certain puzzling findings thrown up by experimental work. This relates in particular to electrical signals generated in smooth muscles, such as synaptic potentials and spikes, and how these are explicable only if three-dimensional syncytial biophysics are taken fully into account. In this talk, I shall provide an illustration of outcomes and insights gleaned from such an approach. I shall first describe our work on the mammalian vas deferens, in which an analysis of the effects of syncytial coupling led us to conclude that the experimental effects of a presumptive gap junction uncoupler, heptanol, on synaptic potentials were incompatible with gap junctional block and could best be explained by a heptanol-induced inhibition of neurotransmitter release, thus compelling a reinterpretation of the mechanism of action of this agent. I shall outline the various lines of evidence, based on indices of syncytial function, that we adduced in order to reach this conclusion. We have now moved on to our current focus on urinary bladder biophysics, where the questions we aim to address are to do with mechanisms of spike generation. Smooth muscle cells in the bladder exhibit spontaneous spiking and spikes occur in a variety of distinct shapes, making their generation problematic to explain. We believe that the variety in shapes may owe less to intrinsic differences in spike mechanism (i.e., in the complement of ion channels participating in spike production) and more to features imposed by syncytial biophysics. We focus especially on the modulation of spike shape in a 3-D coupled network by such factors as innervation pattern, propagation in a syncytium, electrically finite bundles within and between which the spikes spread, and some degree of pacemaker activity by a sub-population of the cells. I shall report two streams of work that we have done, and the tentative conclusions these have enabled us to reach: (a) using the NEURON environment, to construct the smooth muscle syncytium and endow it with synaptic drive, and (b) using signal-processing approaches, towards sorting and classifying the experimentally recorded spikes.


 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
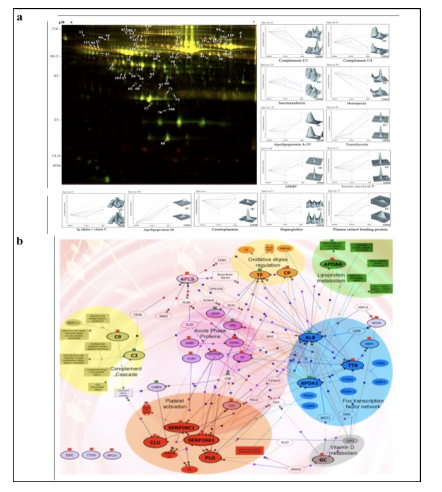
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
![Delegate Talk: Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors @ Sathyam Hall | Vallikavu | Kerala | India](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/bicb.jpg)
Rajasekhar Chekkara, Venkata Reddy Gorla and Sobha Rani Tenkayala
Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors
Polo-like kinase 1 (PLK1) is a significant enzyme with diverse biological actions in cell cycle progression, specifically mitosis. Suppression of PLK1 activity by small molecule inhibitors has been shown to inhibit cancer, being BI 2536 one of the most potent active inhibitor of PLK1 mechanism. Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies were carried out for a set of 54 compounds belonging to Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK1 inhibitors. A six-point pharmacophoremodel AAADDR, with three hydrogen bond acceptors (A), two hydrogen bond donors (D) and one aromatic ring (R) was developed by Phase module of Schrdinger suite Maestro 9. The generated pharmacophore model was used to derive a predictive atom-based 3D quantitative structure-activity relationship analysis (3D-QSAR) model for the training set (r2 = 0.88, SD = 0.21, F = 57.7, N = 44) and for test set (Q2 = 0.51, RMSE = 0.41, PearsonR = 0.79, N = 10). The original set of compounds were docked into the binding site of PLK1 using Glide and the active residues of the binding site were analyzed. The most active compound H18 interacted with active residues Leu 59, Cys133 (glide score = −10.07) and in comparison of BI 2536, which interacted with active residues Leu 59, Cys133 (glide score = −10.02). The 3D-QSAR model suggests that hydrophobic and electron-withdrawing groups are essential for PLK1 inhibitory activity. The docking results describes the hydrogen bond interactions with active residues of these compounds. These results which may support in the design and development of novel PLK1 inhibitors.
 Shrikant Anant, Ph.D.
Shrikant Anant, Ph.D.
The Department of Molecular & Integrative Physiology, Kansas University Medical Center, USA
Cancer Stem Cells: Target Colon Cancers
Shrikant Anant, Deep Kwatra and Dharmalingam Subramaniam
Colon cancer is a leading cause of cancer related deaths in the US, and its rate is increasing at an alarming rate in lndia. Recent studies have suggested the drug resistance role for a mall number of cells within a tumor called cancer stem cells. We identified the colon cancer stem cell marker DCLK1, a member of the protein kinase superfamily and the doublecortin family. The protein encodes a Cterminal serinethreonine protein kinase domain, which shows substantial homology to Ca2calmodulindependent protein kinase. Our current studies have been to identify compounds that can either affect DCLK1 expression or inhibits its activity as a way to inhibit cancer stem cells. Honokiol is a biphenolic compound that has been used in the traditional Chinese Medicine for treating various ailments. In vitro kinase assays with recombinant DCLK1 demonstrated that honokiol inhibits its kinase activity in a dose dependent manner. We therefore determined the effect of honokiol on stem cells. One method to look at effects on stem cells is perform a spheroid assay, where spheroids formation is suggested to maintain stemlike characteristic of cancer cells. Honokiol significantly suppressed colonosphere formation of two colon cancer cell lines HCT116 and SW480. Flow cytometry studies confirmed that honokiol reduced the number of DCLK1cells. A critical signaling pathway known to modulate intestinal stem cell proliferation is the Hippo signaling pathway, and deregulation of the pathway leads to tumor development. DCLK1cells had high levels of YAP1, the nuclear target of Hippo signaling. We determined the effect of honokiol on components of the hipposignaling pathway. Honokiol reduced the phosphorylation of Mst1/2, Lats1/2 and YAP1. Furthermore, honokiol treatment resulted in downregulation of YAPTEAD complex protein TEAD-1. Ectopic expression of the TEAD-1 partially rescued the cells from honokiol mediated growth suppression. To determine the effect of honokiol on tumor growth in vivo, nude mice harboring HCT116 tumor xenografts in their flanks were administered the compound intraperitoneally every day for 21 days. Honokiol treatment significantly inhibited tumor xenograft growth. Western blot and immunohistochemistry analyses demonstrated significant inhibition in the expression of stem marker and Hippo signaling proteins in the honokioltreated xenograft tissues. Taken together, these data suggest that honokiol is a potent inhibitor of colon cancer that targets DCLK1 stem cells by inhibiting Hippo signaling pathway.
 Sudarslal S, Ph.D.
Sudarslal S, Ph.D.
Associate Professor, School of Biotechnology, Amrita University
Electrospray ionization ion trap mass spectrometry for cyclic peptide characterization
There has been considerable interest in the isolation and structural characterization of bioactive peptides produced by bacteria and fungi. Most of the peptides are cyclic depsipeptides characterized by the presence of lactone linkages and β-hydroxy fatty acids. Occurrence of microheterogeneity is another remarkable property of these peptides. Even if tandem mass spectrometers are good analytical tools to structurally characterize peptides and proteins, sequence analysis of cyclic peptides is often ambiguous due to the random ring opening of the peptides and subsequent generation of a set of linear precursor ions with the same m/z. Here we report combined use of chemical derivatization and multistage fragmentation capability of ion trap mass spectrometers to determine primary sequences of a series of closely related cyclic peptides.



Ravindra Gudihal, Suresh Babu C V
Bioanalytical Characterization of Therapeutic Proteins
The characterization of therapeutic proteins such as monoclonal antibody (mAb) during different stages of manufacturing is crucial for timely and successful product release. Regulatory agencies require a variety of analytical technologies for comprehensive and efficient protein analysis. Electrophoresis-based techniques and liquid chromatography (LC) either standalone or coupled to mass spectrometry (MS) are at the forefront for the in-depth analysis of protein purity, isoforms, stability, aggregation, posttranslational modifications, PEGylation, etc. In this presentation, a combination of various chromatographic and electrophoretic techniques such as liquid-phase isoelectric focusing, microfluidic and capillary-based electrophoresis (CE), liquid chromatography (LC) and combinations of those with mass spectrometry techniques will be discussed. We present a workflow based approach to the analysis of therapeutic proteins. In successive steps critical parameters like purity, accurate mass, aggregation, peptide sequence, glycopeptide and glycan analysis are analyzed. In brief, the workflow involved proteolytic digestion of therapeutic protein for peptide mapping, N-Glycanase and chemical labeling reaction for glycan analysis, liquid-phase isoelectric focusing for enrichment of charge variants followed by a very detailed analysis using state of the art methods such as CE-MS and LC-MS. For the analysis of glycans, we use combinations of CE-MS and LC-MS to highlight the sweet spots of these techniques. CE-MS is found to be more useful in analysis of highly sialylated glycans (charged glycans) while nano LC-MS seems to be better adapted for analysis of neutral glycans. These two techniques can be used to get complementary data to profile all the glycans present in a given protein. In addition, microfluidic electrophoresis was used as a QC tool in initial screening for product purity, analysis of papain digestion fragments of mAb, protein PEGylation products, etc. The described workflow involves multiple platforms, provides an end to end solution for comprehensive protein characterization and aims at reducing the total product development time.