 Ganesh Sambasivam, Ph.D.
Ganesh Sambasivam, Ph.D.
CSO & Co-Founder, Anthem Biosciences India
Preclinical Outsourcing to India
The outsourcing segment is witnessing rapid changes with respect to the nature of work outsourced and the location. Cost is the major driver but other considerations such as infrastructure and government policies can also be important drivers for decision making. The last couple of years have been a trying time for all CROs. The global economic meltdown has hit research budgets especially hard. The new challenges facing Contract Research Organizations call for a radically revised approach and a new model that would push the boundaries of this business further and would blur the line between client and vendor further. I believe the term Contract Research Organization (CRO), is a misnomer to begin with (often confused with Clinical Research Organization), has now morphed into a new type of company viz Contract Innovation Services (CIS). Clients are no longer just happy to outsource odds and ends of the development piece but are looking to their vendors for a massive amount of innovation input. This input is increasingly across both the chemistry and discovery domains. This new paradigm calls for CIS companies to develop new platforms, create intellectual propertythat is of service to clients andinnovate processes to meet new found customer expectations.
 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
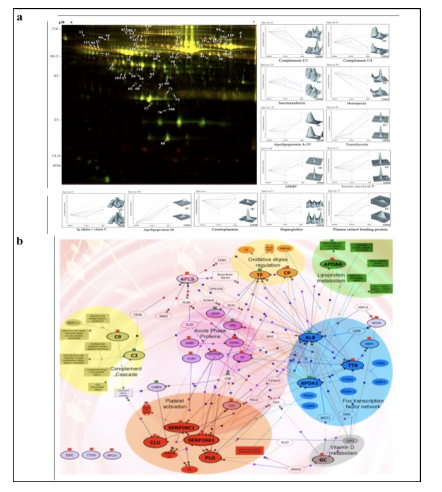
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
 Michelle Hermiston, MD, Ph.D.
Michelle Hermiston, MD, Ph.D.
Assistant Professor, Department of Pediatrics University of California San Francisco, USA
Interrogating Signaling Networks at the Single Cell Level In Primary Human Patient Samples
Multiparameter phosphoflow cytometry is a highly sensitive proteomic approach that enables monitoring of biochemical perturbations at the single cell level. By combining antisera to cell surface markers and key intracellular proteins, perturbations in signaling networks, cell survival and apoptosis mediators, cell cycle regulators, and/or modulators of other cellular processes can be analyzed in a highly reproducible and sensitive manner in the basal state and in response to stimulation or drug treatment. Advantages of this approach include the ability to identify the biochemical consequences of genetic and/or epigenetic changes in small numbers of cells, to map potential interplay between various signaling networks simultaneously in a single cell, and to interrogate potential mechanisms of drug resistance or response in a primary patient sample. Application of this technology to patients with acute lymphoblastic leukemia or the autoimmune disease systemic lupus erythematosus (SLE) will be discussed.
 Srisairam Achuthan, Ph.D.
Srisairam Achuthan, Ph.D.
Senior Scientific Programmer, Research Informatics Division, Department of Information Sciences, City of Hope, CA, USA
Applying Machine learning for Automated Identification of Patient Cohorts
Srisairam Achuthan, Mike Chang, Ajay Shah, Joyce Niland
Patient cohorts for a clinical study are typically identified based on specific selection criteria. In most cases considerable time and effort are spent in finding the most relevant criteria that could potentially lead to a successful study. For complex diseases, this process can be more difficult and error prone since relevant features may not be easily identifiable. Additionally, the information captured in clinical notes is in non-coded text format. Our goal is to discover patterns within the coded and non-coded fields and thereby reveal complex relationships between clinical characteristics across different patients that would be difficult to accomplish manually. Towards this, we have applied machine learning techniques such as artificial neural networks and decision trees to determine patients sharing similar characteristics from available medical records. For this proof of concept study, we used coded and non-coded (i.e., clinical notes) patient data from a clinical database. Coded clinical information such as diagnoses, labs, medications and demographics recorded within the database were pooled together with non-coded information from clinical notes including, smoking status, life style (active / inactive) status derived from clinical notes. The non-coded textual information was identified and interpreted using a Natural Language Processing (NLP) tool I2E from Linguamatics.

Aswath Balakrishnan, Kapaettu Satyamoorthy and Manjunath B Joshi
Introduction
Insulin resistance is a hall mark of metabolic disorders such as diabetes. Reduced insulin response in vasculature leads to disruption of IR/Akt/eNOS signaling pathway resulting in vasoconstriction and subsequently to cardiovascular diseases. Recent studies have demonstrated that inflammatory regulator interleukin-6 (IL-6), as one of the potential mediators that can link chronic inflammation with insulin resistance. Accumulating evidences suggest a significant role of epigenetic mechanisms such as DNA methylation in progression of metabolic disorders. Hence the present study aimed to understand the role of epigenetic mechanisms involved during IL-6 induced vascular insulin resistance and its consequences in cardiovascular diseases.
Materials and Methods
Human umbilical vein endothelial cells (HUVEC) and Human dermal microvascular endothelial cells (HDMEC) were used for this study. Endothelial cells were treated in presence or absence of IL-6 (20ng/ml) for 36 hours and followed by insulin (100nM) stimulation for 15 minutes. Using immunoblotting, cell lysates were stained for phosphor- and total Akt levels to measure insulin resistance. To investigate changes in DNA methylation, cells were treated with or without neutrophil conditioned medium (NCM) as a physiological source of inflammation or IL-6 (at various concentrations) for 36 hours. Genomic DNA was processed for HPLC analysis for methyl cytosine content and cell lysates were analyzed for DNMT1 (DNA (cytosine-5)-methyltransferase 1) and DNMT3A (DNA (cytosine-5)-methyltransferase 3A) levels using immunoblotting.
Results
Endothelial cells stimulated with insulin exhibited an increase in phosphorylation of Aktser 473 in serum free conditions but such insulin response was not observed in cells treated with IL-6, suggesting chronic exposure of endothelial cells to IL-6 leads to insulin resistance. HPLC analysis for global DNA methylation resulted in decreased levels of 5-methyl cytosine in cells treated with pro-inflammatory molecules (both by NCM and IL-6) as compared to untreated controls. Subsequently, analysis in cells treated with IL-6 showed a significant decrease in DNMT1 levels but not in DNMT3A. Other pro-inflammatory marker such as TNF-α did not exhibit such changes.
Conclusion
Our study suggests: a) Chronic treatment of endothelial cells with IL-6 results in insulin resistance b) Neutrophil conditioned medium and IL-6 decreases methyl cytosine levels c) DNMT1 but not DNMT3a levels are reduced in cells treated with IL-6.
 Sudarslal S, Ph.D.
Sudarslal S, Ph.D.
Associate Professor, School of Biotechnology, Amrita University
Electrospray ionization ion trap mass spectrometry for cyclic peptide characterization
There has been considerable interest in the isolation and structural characterization of bioactive peptides produced by bacteria and fungi. Most of the peptides are cyclic depsipeptides characterized by the presence of lactone linkages and β-hydroxy fatty acids. Occurrence of microheterogeneity is another remarkable property of these peptides. Even if tandem mass spectrometers are good analytical tools to structurally characterize peptides and proteins, sequence analysis of cyclic peptides is often ambiguous due to the random ring opening of the peptides and subsequent generation of a set of linear precursor ions with the same m/z. Here we report combined use of chemical derivatization and multistage fragmentation capability of ion trap mass spectrometers to determine primary sequences of a series of closely related cyclic peptides.



Ravindra Gudihal, Suresh Babu C V
Bioanalytical Characterization of Therapeutic Proteins
The characterization of therapeutic proteins such as monoclonal antibody (mAb) during different stages of manufacturing is crucial for timely and successful product release. Regulatory agencies require a variety of analytical technologies for comprehensive and efficient protein analysis. Electrophoresis-based techniques and liquid chromatography (LC) either standalone or coupled to mass spectrometry (MS) are at the forefront for the in-depth analysis of protein purity, isoforms, stability, aggregation, posttranslational modifications, PEGylation, etc. In this presentation, a combination of various chromatographic and electrophoretic techniques such as liquid-phase isoelectric focusing, microfluidic and capillary-based electrophoresis (CE), liquid chromatography (LC) and combinations of those with mass spectrometry techniques will be discussed. We present a workflow based approach to the analysis of therapeutic proteins. In successive steps critical parameters like purity, accurate mass, aggregation, peptide sequence, glycopeptide and glycan analysis are analyzed. In brief, the workflow involved proteolytic digestion of therapeutic protein for peptide mapping, N-Glycanase and chemical labeling reaction for glycan analysis, liquid-phase isoelectric focusing for enrichment of charge variants followed by a very detailed analysis using state of the art methods such as CE-MS and LC-MS. For the analysis of glycans, we use combinations of CE-MS and LC-MS to highlight the sweet spots of these techniques. CE-MS is found to be more useful in analysis of highly sialylated glycans (charged glycans) while nano LC-MS seems to be better adapted for analysis of neutral glycans. These two techniques can be used to get complementary data to profile all the glycans present in a given protein. In addition, microfluidic electrophoresis was used as a QC tool in initial screening for product purity, analysis of papain digestion fragments of mAb, protein PEGylation products, etc. The described workflow involves multiple platforms, provides an end to end solution for comprehensive protein characterization and aims at reducing the total product development time.