

 Pradip K. Bhatnagar, Ph.D.
Pradip K. Bhatnagar, Ph.D.
Former President & Head, Daiichi Sankyo Life Science Research Centre, India
Strategies for Diseases/Target Selection for Drug Discovery and a Multi-Targeted Approach to Metabolic Disorder
Drug discovery and development is a high risk and expensive undertaking. Although, technologies, such as, bioinformatics, genomics, high throughput screening and computer-aided design have helped identify targets, biomarkers, lead candidates and reduced the time required for advancing an idea from bench to clinic, but it still takes 10-12 years and costs approximately one billion dollars to bring a drug to market globally. Therefore, it is imperative that the strategies to reduce the risk and increase efficiency are carefully selected. In this presentation I would discuss strategies for selecting potential diseases, targets and provide an example of multi-targeted approach to metabolic disorder.
 Bharat B. Chattoo, Ph.D.
Bharat B. Chattoo, Ph.D.
Professor, Faculty of Science M.S.University of Baroda, India
Biology of plant infection by Magnaporthe oryzae
The rice blast disease caused by the ascomycetous fungus Magnaporthe oryzae is a major constraint in rice production. Rice-M.oryzae is also emerging as a good model patho-system to investigate how the fungus invades and propagates within the host. Identification and characterisation of genes critical for fungal pathogenesis provides opportunities to explore their use as possible targets for development of strategies for combating fungal infection and to better understand the complex process of host-pathogen interaction.
We have used insertional mutagenesis and RNAi based approaches to identify pathogenesis related genes in this fungus. A large number of mutants were isolated using Agrobacterium tumefaciens mediated transformation (ATMT). Characterisation of several interesting mutants is in progress. We have identified a novel gene, MGA1, required for the development of appressoria. The mutant mga1 is unable to infect and is impaired in glycogen and lipid mobilization required for appressorium development. The glycerol content in the mycelia of the mutant was significantly lower as compared to wild type and it was unable to tolerate hyperosmotic stress. A novel ABC transporter was identified in this fungus. The abc4 mutant did not form functional appressoria, was non-pathogenic and showed increased sensitivity to certain antifungal molecules implying the role of ABC4 in multidrug resistance (MDR). Another mutant MoSUMO (MGG_05737) was isolated using a Split Marker technique; the mutant showed defects in growth, germination and infection. Immuno-fluorescence microscopy revealed that MoSumo is localized to septa in mycelia and nucleus as well as septa in spores. Two Dimensional Gel Electrophoresis showed differences in patterns of protein expression between Wild Type B157 and MoΔSumo mutant. We also isolated and charaterised mutants in MoALR2 (MGG_08843) and MoMNR2 (MGG_09884). Our results indicate that both MoALR2 and MoMNR2 are Mg2+ transporters, and the reduction in the levels of CorA transporters caused defects in surface hydrophobicity, cell wall stress tolerance, sporulation, appressorium formation and infection are mediated through changes in the key signaling cascades in the knock-down transformants. (Work supported by the Department of Biotechnology, Government of India)
 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
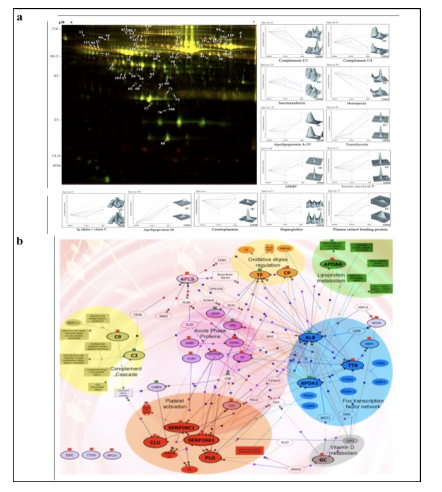
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Suryaprakash Sambhara, DVM, Ph.D
Suryaprakash Sambhara, DVM, Ph.D
Chief, Immunology Section, Influenza Division, CDC, Atlanta, USA
Making sense of pathogen sensors of Innate Immunity: Utility of their ligands as antiviral agents and adjuvants for vaccines.
Currently used antiviral agents act by inhibiting viral entry, replication, or release of viral progeny. However, recent emergence of drug-resistant viruses has become a major public health concern as it is limiting our ability to prevent and treat viral diseases. Furthermore, very few antiviral agents with novel modes of action are currently in development. It is well established that the innate immune system is the first line of defense against invading pathogens. The recognition of diverse pathogen-associated molecular patterns (PAMPs) is accomplished by several classes of pattern recognition receptors (PRRs) and the ligand/receptor interactions trigger an effective innate antiviral response. In the past several years, remarkable progress has been made towards understanding both the structural and functional nature of PAMPs and PRRs. As a result of their indispensable role in virus infection, these ligands have become potential pharmacological agents against viral infections. Since their pathways of action are evolutionarily conserved, the likelihood of viruses developing resistance to PRR activation is diminished. I will discuss the recent developments investigating the potential utility of the ligands of innate immune receptors as antiviral agents and molecular adjuvants for vaccines.



 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
 D. Narasimha Rao, Ph.D.
D. Narasimha Rao, Ph.D.
Professor, Dept of Biochemistry, Indian Institute of Science, Bangalore, India
Genomics of Restriction-Modification Systems
Restriction endonucleases occur ubiquitously among procaryotic organisms. Up to 1% of the genome of procaryotic organisms is taken up by the genes for these enzymes. Their principal biological function is the protection of the host genome against foreign DNA, in particular bacteriophage DNA. Restriction-modification (R-M) systems are composed of pairs of opposing enzyme activities: an endonuclease and a DNA methyltransferase (MTase). The endonucleases recognise specific sequences and catalyse cleavage of double-stranded DNA. The modification MTases catalyse the addition of a methyl group to one nucleotide in each strand of the recognition sequence using S-adenosyl-L-methionine (AdoMet) as the methyl group donor. Based on their molecular structure, sequence recognition, cleavage position and cofactor requirements, R-M systems are generally classified into three groups. In general R-M systems restrict unmodified DNA, but there are other systems that specifically recognise and cut modified DNA. More than 3500 restriction enzymes have been discovered so far. With the identification and sequencing of a number of R-M systems from bacterial genomes, an increasing number of these have been found that do not seem to fit into the conventional classification.
It is well documented that restriction enzyme genes always lie close to their cognate methyltransferase genes. Analysis of the bacterial and archaeal genome sequences shows that MTase genes are more common than one would have expected on the basis of previous biochemical screening. Frequently, they clearly form part of a R-M system, because the adjacent open reading frames (ORFs) show similarity to known restriction enzyme genes. Very often, though, the adjacent ORFs have no homologs in the GenBank and become candidates either for restriction enzymes with novel specificities or for new examples of previously uncloned specificities. Sequence-dependent modification and restriction forms the foundation of defense against foreign DNAs and thus RM systems may serve as a tool of defense for bacterial cells. RM systems however, sometimes behave as discrete units of life, and any threat to their maintenance, such as a challenge by a competing genetic element can lead to cell death through restriction breakage in the genome, thus providing these systems with a competitive advantage. The RM systems can behave as mobile-genetic elements and have undergone extensive horizontal transfer between genomes causing genome rearrangements. The capacity of RM systems to act as selfish, mobile genetic elements may underlie the structure and function of RM enzymes.
The similarities and differences in the different mechanisms used by restriction enzymes will be discussed. Although it is not clear whether the majority of R-M systems are required for the maintenance of the integrity of the genome or whether they are spreading as selfish genetic elements, they are key players in the “genomic metabolism” of procaryotic organisms. As such they deserve the attention of biologists in general. Finally, restriction enzymes are the work horses of molecular biology. Understanding their enzymology will be advantageous to those who use these enzymes, and essential for those who are devoted to the ambitious goal of changing the properties of these enzymes, and thereby make them even more useful.

 Kal Ramnarayan, Ph.D.
Kal Ramnarayan, Ph.D.
Co-founder President & Chief Scientific Officer, Sapient Discovery, San Diego, CA, USA
A cost-effective approach to Protein Structure-guided Drug Discovery: Aided by Bioinformatics, Chemoinformatics and computational chemistry
With the mapping of the human genome completed almost a decade ago, efforts are still underway to understand the gene products (i.e., proteins) in the human biological and disease pathways. Deciphering such information is very important for the discovery and development of small molecule drugs as well as protein therapeutics for various human diseases for which no cure exists. As an example, with more than 500 members, the kinase family of protein targets continues to be an important and attractive class for drug discovery. While how many of the members in this family are actually druggable is still to be established, there are several ongoing efforts on this class of proteins across a broad spectrum of disease categories. Even though in general the protein structural topology might looks similar, there are issues with respect selectivity of identified small molecule inhibitors when, the lead molecule discovery is carried out at the ATP binding site. As an added complexity, allosteric modulators are needed for some of the members, but the actual site for such modulation on the protein target can not resolved with uncertainty. In this presentation we will describe a bioinformatics and computational based platform for small molecule discovery for protein targets that are involved in protein-protein interactions as well as targets like kinases and phosphatases. We will describe a computational approach in which we have used an informatics based platform with several hundred kinases to sort through in silico and identify inhibitors that are likely to be highly selective in the lead generation phase. We will discuss the implication of this approach on the drug discovery of the kinase and phosphatase classes in general and independent of the disease category.

The global healthcare scene of which the pharmaceutical industry and its products are integral components is today at the cross roads. The high and unaffordable costs of drug research with estimates of over 1 billion dollars for every new drug discovered and developed, the very low success rates, the high degree of obsolescence due to undesirable adverse drug reactions, the decline in the development pipeline of new drugs, patent expiries leading to generic competition and the public’s disillusionment with use of chemicals for human consumption as drugs have all significantly contributed to the problems of this lifeline industry. The strategy adopted by the large R&D based Corporations to get bigger and bigger through mergers and acquisitions to improve cost-effectiveness and productivity of R&D has so far not worked effectively. Consequently, one of the recent trends in healthcare, articulated by many experts is to look for alternate or even complementary approaches to reduce the impact of rising costs of drugs on healthcare. Various new strategies for drug discovery such as the use of Natural Products especially medicinal plants are being actively pursued by healthcare planners and providers. Side by side, traditional systems of medicine whether from the oriental countries or the western nations are also having a serious relook to understand their usefulness in healthcare. To achieve its legitimate position in the healthcare scenario, it is essential to scientifically validate their claimed utility through appropriate and systematic research efforts including pre-clinical and clinical studies. In addition to their own use as medicines, knowledge on the Indian Traditional Medicines can be used as a platform for new drug discovery. The huge potential for carrying out systematic R&D programs for new Drug Discovery based on natural products and possible strategies to realise them in the coming decades will be explained in this presentation.

 Ramani A. Aiyer, Ph.D., MBA
Ramani A. Aiyer, Ph.D., MBA
Principal, Shasta BioVentures, San Jose, CA, USA
New Drug R&D in India: Challenges & Opportunities
New drug discovery and development has become a global endeavor, with Western big pharmaceutical companies farming out more and more chemistry and biology research to Asia, particularly India and China. During the last decade, several Indian pharmaceutical companies have embarked on ambitious R&D programs, with slow but steady progress in developing new chemical / molecular entities. The Indian government has also made a strong commitment to promote innovation and entrepreneurship in the biotechnology sector. The first part of the talk will focus on a case study showing the entire process of discovery and development of a new drug recently launched for Rheumatoid Arthritis. We will then address the challenges of conducting innovative R&D in India and actions necessary to overcome them. The second part of the talk will make the case for developing Ayurvedic drug formulations for the Western / Global markets, again using the example of Rheumatoid Arthritis (Aamavaata). Ayurveda takes a holistic approach to disease diagnosis and therapy based on interactions among body type (prakriti), tri-doshas (three body humors), sapta-dhatus (seven tissues) and malas (excretions). The drugs prescribed are usually herbo-mineral formulations comprising multiple medicinal plants and / or metals. The manufacturing processes date back to Ayurvedic texts several thousand years old, and are compiled in the Ayurvedic Pharmacopeia. Also, the treatment modalities and drug formulations are “personalized” to fit different patient types, based on the holistic diagnoses mentioned earlier. There is a tremendous need to establish a sound basis for Ayurvedic drug discovery R&D for the modern world. We must find a scientific and ethical way to leverage the vast body of anecdotal and possibly retrospective data on patients undergoing Ayurvedic treatment. Combined with in vitro and in vivo biological data on Ayurvedic herbo-mineral formulations, the adoption of stringent manufacturing practices, and designing sound clinical trials to establish the safety and efficacy, India has a golden opportunity to expand the reach of Ayurvedic drugs into Western / Global medical practice.


Tejaswini Subbannayya, Nandini A. Sahasrabuddhe, Arivusudar Marimuthu, Santosh Renuse, Gajanan Sathe, Srinivas M. Srikanth, Mustafa A. Barbhuiya, Bipin Nair, Juan Carlos Roa, Rafael Guerrero-Preston, H. C. Harsha, David Sidransky, Akhilesh Pandey, T. S. Keshava Prasad and Aditi Chatterjee
Proteomic profiling of gallbladder cancer secretome – a source for circulatory biomarker discovery
Gallbladder cancer (GBC) is the fifth most common cancer of the gastrointestinal tract and one of the common malignancies that occur in the biliary tract (Misra et al. 2006; Lazcano-Ponce et al. 2001). It has a poor prognosis with survival of less than 5 years in 90% of the cases (Misra et al. 2003). The etiology is ill-defined. Several risk factors have been reported including cholelithiasis, obesity, female gender and exposure to carcinogens (Eslick 2010; Kumar et al. 2006). Poor prognosis in GBC is mainly due to late presentation of the disease and lack of reliable biomarkers for early diagnosis. This emphasizes the need to identify and characterize cancer biomarkers to aid in the diagnosis and prognosis of GBC. Secreted proteins are an important class of molecules which can be detected in body fluids and has been targeted for biomarker discovery. There are challenges faced in the proteomic interrogation of body fluids especially plasma such as low abundance of tumor secreted proteins, high complexity and high abundance of other proteins that are not released by the tumor cells (Tonack et al. 2009). Profiling of conditioned media from the cancer cell lines can be used as an alternate means to identify secreted proteins from tumor cells (Kashyap et al. 2010; Marimuthu et al. 2012). We analyzed the invasive property of 7 GBC cell lines (SNU-308, G-415, GB-d1, TGBC2TKB, TGBC24TKB, OCUG-1 and NOZ). Four cell lines were selected for analysis of the cancer secretome based on the invasive property of the cells. We employed isobaric tags for relative and absolute quantitation (iTRAQ) labeling technology coupled with high resolution mass spectrometry to identify and characterize secretome from the panel of 4GBC cancer cells mentioned above. In total, we have identified around 2,000 proteins of which 175 were secreted at differential abundance across all the four cell lines. This secretome analysis will act as a reservoir of candidate biomarkers. Currently, we are investigating and validating these candidate markers from GBC cell secretome. Through this study, we have shown mass spectrometry-based quantitative proteomic analysis as a robust approach to investigate secreted proteins in cancer cells.