


 Gaute Einevoll, Ph.D.
Gaute Einevoll, Ph.D.
Professor of Physics, Department of Mathematical Sciences & Technology, Norwegian University of Life Sciences (UMB)
Multiscale modeling of cortical network activity: Key challenges
Gaute T. Einevoll Computational Neuroscience Group, Norwegian University of Life Sciences, 1432 Ås, Norway; Norwegian National Node of the International Neuroinformatics Coordinating Facility (INCF)
Several challenges must be met in the development of multiscale models of cortical network activity. In the presentation I will, based on ongoing work in our group (http://compneuro.umb.no/ ) on multiscale modeling of cortical columns, outline some of them. In particular,
-
Combined modeling schemes for neuronal, glial and vascular dynamics [1,2],
-
Development of sets of interconnected models describing system at different levels of biophysical detail [3-5],
-
Multimodal modeling, i.e., how to model what you can measure [6-12],
-
How to model when you don’t know all the parameters, and
-
Development of neuroinformatics tools and routines to do simulations efficiently and accurately [13,14].
References:
- L. Øyehaug, I. Østby, C. Lloyd, S.W. Omholt, and G.T. Einevoll: Dependence of spontaneous neuronal firing and depolarisation block on astroglial membrane transport mechanisms, J Comput Neurosci 32, 147-165 (2012)
- I. Østby, L. Øyehaug, G.T. Einevoll, E. Nagelhus, E. Plahte, T. Zeuthen, C. Lloyd, O.P. Ottersen, and S.W. Omholt: Astrocytic mechanisms explaining neural-activity-induced shrinkage of extraneuronal space, PLoS Comp Biol 5, e1000272 (2009)
- T. Heiberg, B. Kriener, T. Tetzlaff, A. Casti, G.T. Einevoll, and H.E. Plesser: Firing-rate models can describe the dynamics of the retina-LGN connection, J Comput Neurosci (2013)
- T. Tetzlaff, M. Helias, G.T. Einevoll, and M. Diesmann: Decorrelation of neural-network activity by inhibitory feedback, PLoS Comp Biol 8, e10002596 (2012).
- E. Nordlie, T. Tetzlaff, and G.T. Einevoll: Rate dynamics of leaky integrate-and-fire neurons with strong synapses, Frontiers in Comput Neurosci 4, 149 (2010)
- G.T. Einevoll, F. Franke, E. Hagen, C. Pouzat, K.D. Harris: Towards reliable spike-train recording from thousands of neurons with multielectrodes, Current Opinion in Neurobiology 22, 11-17 (2012)
- H. Linden, T Tetzlaff, TC Potjans, KH Pettersen, S Grun, M Diesmann, GT Einevoll: Modeling the spatial reach of LFP, Neuron 72, 859-872 (2011).
- H. Linden, K.H. Pettersen, and G.T. Einevoll: Intrinsic dendritic filtering gives low-pass power spectra of local field potentials, J Computational Neurosci 29, 423-444 (2010)
- K.H. Pettersen and G.T. Einevoll: Amplitude variability and extracellular low-pass filtering of neuronal spikes, Biophysical Journal 94, 784-802 (2008).
- K.H. Pettersen, E. Hagen, and G.T. Einevoll: Estimation of population firing rates and current source densities from laminar electrode recordings, J Comput Neurosci 24, 291-313 (2008).
- K. Pettersen, A. Devor, I. Ulbert, A.M. Dale and G.T. Einevoll. Current-source density estimation based on inversion of electrostatic forward solution: Effects of finite extent of neuronal activity and conductivity discontinuities, Journal of Neuroscience Methods 154, 116-133 (2006).
- G.T. Einevoll, K. Pettersen, A. Devor, I. Ulbert, E. Halgren and A.M. Dale: Laminar Population Analysis: Estimating firing rates and evoked synaptic activity from multielectrode recordings in rat barrel cortex, Journal of Neurophysiology 97, 2174-2190 (2007).
- LFPy: A tool for simulation of extracellular potentials (http://compneuro.umb.no)
- E. Nordlie, M.-O. Gewaltig, H. E. Plesser: Towards reproducible descriptions of neuronal network models, PLoS Comp Biol 5, e1000456 (2009).

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
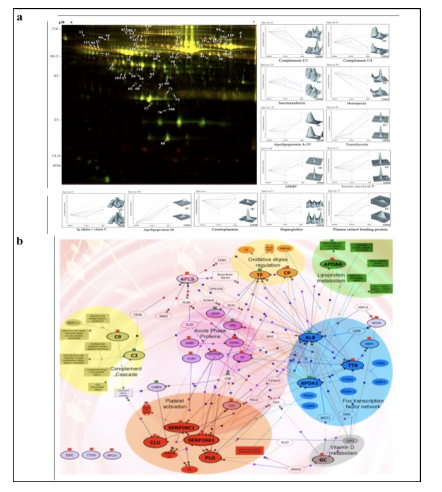
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
 R. Manjunath, Ph.D.
R. Manjunath, Ph.D.
Associate Professor, Dept of Biochemistry, Indian Institute of Science, Bengaluru, India
REGULATION OF THE MHC COMPLEX AND HLA SOLUBILISATION BY THE FLAVIVIRUS, JAPANESE ENCEPHALITIS VIRUS
Viral encephalitis caused by Japanese encephalitis virus (JEV) and West Nile Virus (WNV) is a mosquito-borne disease that is prevalent in different parts of India and other parts of South East Asia. JEV is a positive single stranded RNA virus that belongs to the Flavivirus genus of the family Flaviviridae. The genome of JEV is about 11 kb long and codes for a polyprotein which is cleaved by both host and viral encoded proteases to form 3 structural and 7 non-structural proteins. It is a neurotropic virus which infects the central nervous system (CNS) and causes death predominantly in newborn children and young adults. JEV follows a zoonotic life-cycle involving mosquitoes and vertebrate, chiefly pigs and ardeid birds, as amplifying hosts. Humans are infected when bitten by an infected mosquito and are dead end hosts. Its structural, pathological, immunological and epidemiological aspects have been well studied. After entry into the host following a mosquito bite, JEV infection leads to acute peripheral neutrophil leucocytosis in the brain and leads to elevated levels of type I interferon, macrophage-derived chemotactic factor, RANTES,TNF-α and IL-8 in the serum and cerebrospinal fluid.
Major Histocompatibility Complex (MHC) molecules play a very important role in adaptive immune responses. Along with various classical MHC class I molecules, other non-classical MHC class I molecules play an important role in modulating innate immune responses. Our lab has shown the activation of cytotoxic T-cells (CTLs) during JEV infection and CTLs recognize non-self peptides presented on MHC molecules and provide protection by eliminating infected cells. However, along with proinflammatory cytokines such as TNFα, they may also cause immunopathology within the JEV infected brain. Both JEV and WNV, another related flavivirus have been shown to increase MHC class I expression. Infection of human foreskin fibroblast cells (HFF) by WNV results in upregulation of HLA expression. Data from our lab has also shown that JEV infection upregulates classical as well as nonclassical (class Ib) MHC antigen expression on the surface of primary mouse brain astrocytes and mouse embryonic fibroblasts.
There are no reports that have discussed the expression of these molecules on other cells like endothelial and astrocyte that play an important role in viral invasion in humans. We have studied the expression of human classical class I molecules HLA-A, -B, -C and the non-classical HLA molecules, HLA-E as well as HLA-F in immortalized human brain microvascular endothelial cells (HBMEC), human endothelial cell line (ECV304), human glioblastoma cell line (U87MG) and human foreskin fibroblast cells (HFF). Nonclassical MHC molecules such as mouse Qa-1b and its human homologue, HLA-E have been shown to be the ligand for the inhibitory NK receptor, NKG2A/CD94 and may bridge innate and adaptive immune responses. We show that JEV infection of HBMEC and ECV 304 cells upregulates the expression of HLA-A, and –B antigens as well as HLA-E and HLA-F. Increased expression of total HLA-E upon JEV infection was also observed in other human cell lines as well like, human amniotic epithelial cells, AV-3, FL and WISH cells. Further, we show for the first time that soluble HLA-E (sHLA-E) was released from infected ECV and HBMECs. In contrast, HFF cells showed only upregulation of cell-surface HLA-E expression while U87MG, a human glioblastoma cell line neither showed any cell-surface induction nor its solubilization. This shedding of sHLA-E was found to be dependent on matrix metalloproteinase (MMP) and an important MMP, MMP-9 was upregulated during JEV infection. Treatment with IFNγ resulted in the shedding of sHLA-E from ECV as well as U87MG but not from HFF cells. Also, sHLA-E was shed upon treatment with IFNβ and both IFNβ and TNFα, when present together caused an additive increase in the shedding of sHLA-E. HLA-E is an inhibitory ligand for CD94/NKG2A receptor of Natural Killer cells. Thus, MMP mediated solubilization of HLA-E from infected endothelial cells may have important implications in JEV pathogenesis including its ability to compromise the blood brain barrier.

 Lalitha Subramanian, Ph.D.
Lalitha Subramanian, Ph.D.
Chief Scientific Officer & VP, Services at Scienomics, USA
Nanoscale Simulations – Tackling Form and Formulation Challenges in Drug Development and Drug Delivery
Lalitha Subramanian, Dora Spyriouni, Andreas Bick, Sabine Schweizer, and Xenophon Krokidis Scienomics
The discovery of a compound which is potent in activity against a target is a major milestone in Pharmaceutical and Biotech industry. However, a potent compound is only effective as a therapeutic agent when it can be administered such that the optimal quantity is transported to the site of action at an optimal rate. The active pharmaceutical ingredient (API) has to be tested for its physicochemical properties before the appropriate dosage form and formulation can be designed. Some of the commonly evaluated parameters are crystal forms and polymorphs, solubility, dissolution behavior, stability, partition coefficient, water sorption behavior, surface properties, particle size and shape, etc. Pharmaceutical development teams face the challenge of quickly and efficiently determining a number of properties with small quantities of the expensive candidate compounds. Recently the trend has been to screen these properties as early as possible and often the candidate compounds are not available in sufficient quantities. Increasingly, these teams are leveraging nanoscale simulations similar to those employed by drug discovery teams for several decades. Nanoscale simulations are used to predict the behavior using very little experimental data and only if this is promising further experiments are done. Another aspect where nanoscale simulations are being used in drug development and drug delivery is to get insights into the behavior of the system so that process failures can be remediated and formulation performance can be improved. Thus, the predictive screening and the in-depth understanding leads to experimental efficiency resulting in far-reaching business impacts.
With specific examples, this talk will focus on the different types of nanoscale simulations used to predict properties of the API in excipients and also provide insight into system behavior as a function of shelf life, temperature, mechanical stress, etc.
 Srisairam Achuthan, Ph.D.
Srisairam Achuthan, Ph.D.
Senior Scientific Programmer, Research Informatics Division, Department of Information Sciences, City of Hope, CA, USA
Applying Machine learning for Automated Identification of Patient Cohorts
Srisairam Achuthan, Mike Chang, Ajay Shah, Joyce Niland
Patient cohorts for a clinical study are typically identified based on specific selection criteria. In most cases considerable time and effort are spent in finding the most relevant criteria that could potentially lead to a successful study. For complex diseases, this process can be more difficult and error prone since relevant features may not be easily identifiable. Additionally, the information captured in clinical notes is in non-coded text format. Our goal is to discover patterns within the coded and non-coded fields and thereby reveal complex relationships between clinical characteristics across different patients that would be difficult to accomplish manually. Towards this, we have applied machine learning techniques such as artificial neural networks and decision trees to determine patients sharing similar characteristics from available medical records. For this proof of concept study, we used coded and non-coded (i.e., clinical notes) patient data from a clinical database. Coded clinical information such as diagnoses, labs, medications and demographics recorded within the database were pooled together with non-coded information from clinical notes including, smoking status, life style (active / inactive) status derived from clinical notes. The non-coded textual information was identified and interpreted using a Natural Language Processing (NLP) tool I2E from Linguamatics.