 Gillian Murphy, Ph.D.
Gillian Murphy, Ph.D.
Professor, Department of Oncology, University of Cambridge, UK
A novel strategy for targeting metalloproteinases in cancer
Epithelial tumours evolve in a multi-step manner, involving both inflammatory and mesenchymal cells. Although intrinsic factors drive malignant progression, the influence of the micro-environment of neoplastic cells is a major feature of tumorigenesis. Extracellular proteinases, notably the metalloproteinases, are key players in the regulation of this cellular environment, acting as major effectors of both cell-cell and cell-extracellular matrix (ECM) interactions. They are involved in modifying ECM integrity, growth factor availability and the function of cell surface signalling systems, with consequent effects on cellular differentiation, proliferation and apoptosis.This has made metalloproteinases important targets for therapeutic interventions in cancer and small molecule inhibitors focussed on chelation of the active site zinc and binding within the immediate active site pocket were developed. These were not successful in early clinical trials due to the relative lack of specificity and precise knowledge of the target proteinase(s) in specific cancers. We can now appreciate that it is essential that we understand the relative roles of the different enzymes (of which there are over 60) in terms of their pro and anti tumour activity and their precise sites of expression The next generations of metalloproteinase inhibitors need the added specificity that might be gained from an understanding of the structure of individual active sites and the role of extra catalytic domains in substrate binding and other aspects of their biology. We have prepared scFv antibodies to the extra catalytic domains of two membrane metalloproteinases, MMP-14 and ADAM17, that play key roles in the tumour microenvironment. Our rationale and experiences with these agents will be presented in more detail.

 Colin Barrow, Ph.D.
Colin Barrow, Ph.D.
Chair in Biotechnology, School of Life & Environmental Sciences, Deakin University, Australia
Nano-biotechnology: Omega-3 Oils and Nanofibres
The health benefits of long-chain omega-3 fatty acids are well established, especially for eicosapentaenoic acid (EPA) and docosapentaenoic acid (DHA) from fish and microbial sources. In fact, a billion dollar market exists for these compounds as nutritional supplements, functional foods and pharmaceuticals. This presentation will describe some aspects of our omega-3 biotechnology research that are at the intersection of Nano-biotechnology and oil chemistry. These include the use of lipases for the concentration of omega-3 fats, through immobilization of these lipases on nanoparticles, and the microencapsulation and stabilization of omega-3 oils for functional foods. I will also describe some of our work on the enzymatic production of resolvins using lipoxygenases, and the fermentation of omega-3 oils from marine micro-organisms. Finally, I will describe some of our work on the formation of amyloid fibrils and graphene for various applications in nano-biotechnology.
 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
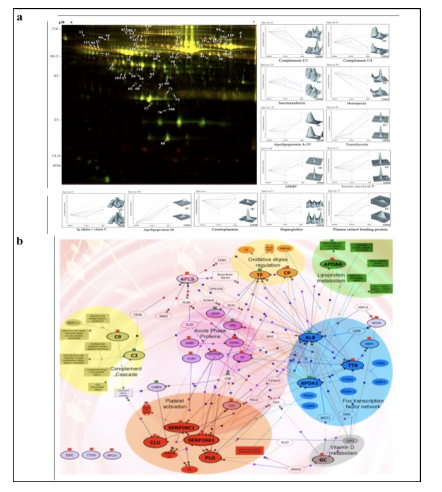
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)


 Jaap Heringa, Ph.D.
Jaap Heringa, Ph.D.
Director & Professor of Bioinformatics, IBIVU VU University Amsterdam, The Netherlands
Modeling strategy based on Petri-nets
In my talk I will introduce a formal modeling strategy based on Petri-nets, which are a convenient means of modeling biological processes. I will illustrate the capabilities of Petri-nets as reasoning vehicles using two examples: Haematopoietic stem cell differentiation in mice, and vulval development in C. elegance. The first system was modeled using a Boolean implementation, and the second using a coarse-grained multi-cellular Petri-net model. Concepts such as the model state space, attractor states, and reasoning to adapt the model to the biological reality will be discussed.

 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
 D. Narasimha Rao, Ph.D.
D. Narasimha Rao, Ph.D.
Professor, Dept of Biochemistry, Indian Institute of Science, Bangalore, India
Genomics of Restriction-Modification Systems
Restriction endonucleases occur ubiquitously among procaryotic organisms. Up to 1% of the genome of procaryotic organisms is taken up by the genes for these enzymes. Their principal biological function is the protection of the host genome against foreign DNA, in particular bacteriophage DNA. Restriction-modification (R-M) systems are composed of pairs of opposing enzyme activities: an endonuclease and a DNA methyltransferase (MTase). The endonucleases recognise specific sequences and catalyse cleavage of double-stranded DNA. The modification MTases catalyse the addition of a methyl group to one nucleotide in each strand of the recognition sequence using S-adenosyl-L-methionine (AdoMet) as the methyl group donor. Based on their molecular structure, sequence recognition, cleavage position and cofactor requirements, R-M systems are generally classified into three groups. In general R-M systems restrict unmodified DNA, but there are other systems that specifically recognise and cut modified DNA. More than 3500 restriction enzymes have been discovered so far. With the identification and sequencing of a number of R-M systems from bacterial genomes, an increasing number of these have been found that do not seem to fit into the conventional classification.
It is well documented that restriction enzyme genes always lie close to their cognate methyltransferase genes. Analysis of the bacterial and archaeal genome sequences shows that MTase genes are more common than one would have expected on the basis of previous biochemical screening. Frequently, they clearly form part of a R-M system, because the adjacent open reading frames (ORFs) show similarity to known restriction enzyme genes. Very often, though, the adjacent ORFs have no homologs in the GenBank and become candidates either for restriction enzymes with novel specificities or for new examples of previously uncloned specificities. Sequence-dependent modification and restriction forms the foundation of defense against foreign DNAs and thus RM systems may serve as a tool of defense for bacterial cells. RM systems however, sometimes behave as discrete units of life, and any threat to their maintenance, such as a challenge by a competing genetic element can lead to cell death through restriction breakage in the genome, thus providing these systems with a competitive advantage. The RM systems can behave as mobile-genetic elements and have undergone extensive horizontal transfer between genomes causing genome rearrangements. The capacity of RM systems to act as selfish, mobile genetic elements may underlie the structure and function of RM enzymes.
The similarities and differences in the different mechanisms used by restriction enzymes will be discussed. Although it is not clear whether the majority of R-M systems are required for the maintenance of the integrity of the genome or whether they are spreading as selfish genetic elements, they are key players in the “genomic metabolism” of procaryotic organisms. As such they deserve the attention of biologists in general. Finally, restriction enzymes are the work horses of molecular biology. Understanding their enzymology will be advantageous to those who use these enzymes, and essential for those who are devoted to the ambitious goal of changing the properties of these enzymes, and thereby make them even more useful.

 Satheesh Babu T. G., Ph.D.
Satheesh Babu T. G., Ph.D.
Associate Professor, Department of Sciences, School of Engineering, Amrita University, Coimbatore, India
Nanomaterials for ‘enzyme-free’ biosensing
Enzyme based sensors have many draw backs such as poor storage stability, easily affected by the change in pH and temperature and involves complicated enzyme immobilization procedures. To address this limitation, an alternative approach without the use of enzyme, “non-enzymatic” has been tried recently. Choosing the right catalyst for direct electrochemical oxidation / reduction of a target molecule is the key step in the fabrication of non-enzymatic sensors.
Non-enzymatic sensors for glucose, creatinine, vitamins and cholesterol are fabricated using different nanomaterials, such as nanotubes, nanowires and nanoparticles of copper oxide, titanium dioxide, tantalum oxide, platinum, gold and graphenes. These sensors selectively catalyse the targeted analyte with very high sensitivity. These nanomaterials based sensors combat the drawbacks of enzymatic sensors.

![Delegate Talk: Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors @ Sathyam Hall | Vallikavu | Kerala | India](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/bicb.jpg)
Rajasekhar Chekkara, Venkata Reddy Gorla and Sobha Rani Tenkayala
Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies on Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK 1 inhibitors
Polo-like kinase 1 (PLK1) is a significant enzyme with diverse biological actions in cell cycle progression, specifically mitosis. Suppression of PLK1 activity by small molecule inhibitors has been shown to inhibit cancer, being BI 2536 one of the most potent active inhibitor of PLK1 mechanism. Pharmacophore modeling, atom-based 3D-QSAR and molecular docking studies were carried out for a set of 54 compounds belonging to Pyrimido[5,4-e][1,2,4]triazine derivatives as PLK1 inhibitors. A six-point pharmacophoremodel AAADDR, with three hydrogen bond acceptors (A), two hydrogen bond donors (D) and one aromatic ring (R) was developed by Phase module of Schrdinger suite Maestro 9. The generated pharmacophore model was used to derive a predictive atom-based 3D quantitative structure-activity relationship analysis (3D-QSAR) model for the training set (r2 = 0.88, SD = 0.21, F = 57.7, N = 44) and for test set (Q2 = 0.51, RMSE = 0.41, PearsonR = 0.79, N = 10). The original set of compounds were docked into the binding site of PLK1 using Glide and the active residues of the binding site were analyzed. The most active compound H18 interacted with active residues Leu 59, Cys133 (glide score = −10.07) and in comparison of BI 2536, which interacted with active residues Leu 59, Cys133 (glide score = −10.02). The 3D-QSAR model suggests that hydrophobic and electron-withdrawing groups are essential for PLK1 inhibitory activity. The docking results describes the hydrogen bond interactions with active residues of these compounds. These results which may support in the design and development of novel PLK1 inhibitors.

Anupama Natarajan, James Hickman and Peter Molnar
Novel Cell-Based Biosensors for High Throughput Toxin Detection and Drug Screening Applications
Over the last decade there has been focus on the development of cellbased biosensors to detect environmental toxins or to combat the threats of biological warfare. These sensors have been shown to have multiple applications including understanding function and behaviour at the cellular and tissue levels, in cell electrophysiology and as drug screening tools that can eliminate animal testing. These factors make the development of cell-based biosensors into high throughput systems a priority in pharmacological, environmental and defence industries (Pancrazio J J et al. 1999, Kang G et al. 2009, Krinke D et al. 2009). We have developed a high through-put in vitro cell-silicon hybrid platform that could be used to analyze both cell function and response to various toxins and drugs. Our hypothesis was that by utilizing surface modification to provide external guidance cues as well as optimal growth conditions for different cell types (Cardiac and Neuronal), we could enhance the information output and content of such a system. An intrinsic part of this study was to create ordered or patterned functional networks of cells on Micro-electrode arrays (MEA). Such engineered networks had a two-fold purpose in that they not only aided in a more accurate analysis of cell response and cell and tissue behaviour, but also increased the efficiency of the system by increasing the connectivity and placement of the cells over the recording electrodes. Here we show the response of this system to various toxins and drugs and the measurement of several vital cardiac parameters like conduction velocity and refractory period (Natarajan A et al. 2011)
 Jeff Perry, Ph.D.
Jeff Perry, Ph.D.
Assistant Professor, University of California, Riverside
Combined Crystallography and SAXS Methods for Studying Macromolecular Complexes
Recent developments in small angle X-ray scattering (SAXS) are rapidly providing new insights into protein interactions, complexes and conformational states in solution, allowing for detailed biophysical quantification of samples of interest1. Initial analyses provide a judgment of sample quality, revealing the potential presence of aggregation, the overall extent of folding or disorder, the radius of gyration, maximum particle dimensions and oligomerization state. Structural characterizations may include ab initio approaches from SAXS data alone, or enhance structural solutions when combined with previously determined crystal/NMR domains. This combination can provide definitions of architectures, spatial organizations of the protein domains within a complex, including those not yet determined by crystallography or NMR, as well as defining key conformational states. Advantageously, SAXS is not generally constrained by macromolecule size, and rapid collection of data in a 96-well plate format provides methods to screen sample conditions. Such screens include co-factors, substrates, differing protein or nucleotide partners or small molecule inhibitors, to more fully characterize the variations within assembly states and key conformational changes. These analyses are also useful for screening constructs and conditions that are most likely to promote crystal growth. Moreover, these high throughput structural determinations can be leveraged to define how polymorphisms affect assembly formations and activities. Also, SAXS-based technologies may be potentially used for novel structure-based screening, for compounds inducing shape changes or associations/diassociations. This is addition to defining architectural characterizations of complexes and interactions for systems biology-based research, and distinctions in assemblies and interactions in comparative genomics. Thus, SAXS combined with crystallography/NMR and computation provides a unique set of tools that should be considered as being part of one’s repertoire of biophysical analyses, when conducting characterizations of protein and other macromolecular interactions.
1 Perry JJ & Tainer JA. Developing advanced X-ray scattering methods combined with crystallography and computation. Methods. 2013 Mar;59(3):363-71.
