 Ayyappan Nair, Ph.D.
Ayyappan Nair, Ph.D.
Head, Business Development (Technologies, Discovery Biology), Anthem Biosciences & DavosPharma, New Jersey, USA
Inhibition of NF-κB regulated gene expression by chrysoeriol suppresses tumorigenesis in breast cancer cells
Amrutha K1, Pandurangan Nanjan1, Sanu K Shaji1, Damu Sunilkumar1, Subhalakshmi K1, Rashmi U Nair1, Lakshmi Rajakrishna2, Asoke Banerji1, Ayyappan Ramesh Nair1*,2
- School of Biotechnology, Amrita Vishwa Vidyapeetham, Amritapuri Campus, Clappana P.O., Kollam – 690 525, Kerala, India
- Anthem Biosciences, No 49, Canara Bank Road, Bommasandra Industrial Area, Phase 1, Hosur Road, Bangalore – 560 099, Karnataka, India
Abstract: A large number of effective cancer-preventing compounds inhibit the activation of nuclear factor-κ B (NF-κB). It has been previously demonstrated that some flavonoids that are a vital component of our diet inhibits this pathway. As a consequence, many flavonoids inhibit genes involved in various aspects of tumorigenesis and have thus emerged as potential chemopreventive candidates for cancer treatment. We studied the effect of 17 different flavonoids, including the highly evaluated quercetin on the NF-κB pathway, and on the expression of MMP-9 and COX-2 (two NF-κB regulated genes involved in metastasis) in the highly invasive human breast cancer cell line MDA-MB-231. The findings suggest that not all the quercetin like flavone backbone compounds inhibit the NF-κB pathway, and that the highly hydoxylated flavonols quercetagetin and gossypetin did not inhibit this pathway, nor did it inhibit the expression of MMP-9 and COX-2. This indicates a correlation between inhibition of NF-κB and subsequent suppression of these NF-κB regulated genes. Here, we also report the novel observation that the not so well characterized methoxylated flavone chrysoeriol inhibited the NF-κB pathway, and was most potent in reducing the expression of MMP-9 and COX-2. Based on these observations, the cellular effects of chrysoeriol were evaluated in MDA-MB-231. Chrysoeriol caused cell cycle arrest at G2/M, inhibited migration and invasion, and caused cell death of macrophages that contributed to migration of these cancer cells. These effects of chrysoeriol make it a potential therapeutic candidate for breast cancer metastasis.

 Sanjeeva Srivastava, Ph.D.
Sanjeeva Srivastava, Ph.D.
Assistant Professor, Proteomics Lab, IIT-Bombay, India
Identification of Potential Early Diagnostic Biomarkers for Gliomas and Various Infectious Diseases using Proteomic Technologies
The spectacular advancements achieved in the field of proteomics research during the last decade have propelled the growth of proteomics for clinical research. Recently, comprehensive proteomic analyses of different biological samples such as serum or plasma, tissue, CSF, urine, saliva etc. have attracted considerable attention for the identification of protein biomarkers as early detection surrogates for diseases (Ray et al., 2011). Biomarkers are biomolecules that can be used for early disease detection, differentiation between closely related diseases with similar clinical manifestations as well as aid in scrutinizing disease progression. Our research group is performing in-depth analysis of alteration in human proteome in different types of brain tumors and various pathogenic infections to obtain mechanistic insight about the disease pathogenesis and host immune responses, and identification of surrogate protein markers for these fatal human diseases.
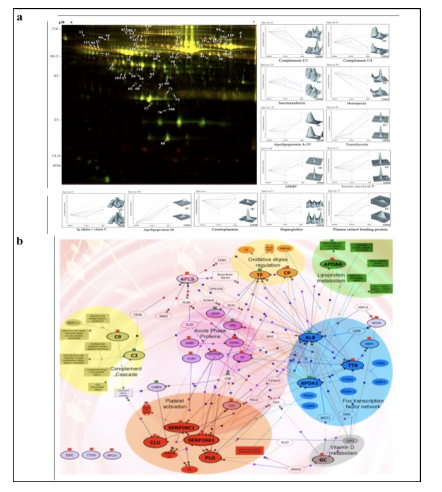
Applying 2D-DIGE in combination with MALDI-TOF/TOF MS we have analyzed the serum and tissue proteome profiles of glioblastoma multiforme; the most common and lethal adult malignant brain tumor (Gollapalli et al., 2012) (Figure 1). Results obtained were validated by employing different immunoassay-based approaches. In serum proteomic analysis we have identified some interesting proteins like haptoglobin, ceruloplasmin, vitamin-D binding protein etc. Moreover, proteomic analysis of different grades (grade-I to IV) of gliomas and normal brain tissue was performed and differential expressions of quite a few proteins such as SIRT2, GFAP, SOD, CDC42 have been identified, which have significant correlation with the tumor growth. While proteomic analysis of cerebrospinal fluid from low grade (grade I & II) vs. high grade (grade III & IV) gliomas revealed modulation of CSF levels of apolipoprotein E, dickkopf related protein 3, vitamin D binding protein and albumin in high grade gliomas. The prospective candidates identified in our studies provide a mechanistic insight of glioma pathogenesis and identification of potential biomarkers. We are also studying the role of JAK/STAT interactome and therapeutic potential of STAT3 inhibitors in gliomas using proteomics approach. Several candidates of the JAK/STAT interactome were identified with altered expression and a significant correlation was observed between STAT3 and PDK1 transcript expression level.
We have also investigated the changes in human serum proteome in different infectious diseases including falciparum and vivax malaria (Ray et al., 2012a; Ray et al., 2012b), dengue (Ray et al., 2012c) and leptospirosis (Srivastava et al., 2012). Although, quite a few serum proteins were found to be commonly altered in different infectious diseases and might be a consequence of inflammation mediated acute phase response signaling, uniquely modulated candidates were identified in each pathogenic infection indicating the some inimitable responses. Further, a panel of identified proteins consists of six candidates; serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I was used to build statistical sample class prediction models employing PLSDA and other classification methods to predict the clinical phenotypic classes and 91.37% overall prediction accuracy was achieved (Figure 2). ROC curve analysis was carried out to evaluate the individual performance of classifier proteins. The excellent discrimination among the different disease groups on the basis of differentially expressed proteins demonstrates the potential diagnostic implications of this analytical approach.
Keywords: Diagnostic biomarkers, Gliomas, Infectious Diseases, Proteomics, Serum proteome
Acknowledgments: This disease biomarker discovery research was supported by Department of Biotechnology, India grant (No. BT/PR14359/MED/30/916/2010), Board of Research in Nuclear Sciences (BRNS) DAE young scientist award (2009/20/37/4/BRNS) and a startup grant 09IRCC007 from the IIT Bombay. The active support from Advanced Center for Treatment Research and Education in Cancer (ACTREC), Tata Memorial Hospital (TMH), and Seth GS Medical College and KEM Hospital Mumbai, India in clinical sample collection process is gratefully acknowledged.
References :
- Ray S, Reddy PJ, Jain R, Gollapalli K. Moiyadi A, Srivastava S. Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 11: 2139-61, 2011.
- Gollapalli K, Ray S, Srivastava R, Renu D, Singh P, Dhali S, Dikshit JB, Srikanth R, Moiyadi A, Srivastava S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 12(14): 2378-90, 2012.
- Ray S, Renu D, Srivastava R, Gollapalli K, Taur S, Jhaveri T, Dhali S, Chennareddy S, Potla A, Dikshit JB, Srikanth R, Gogtay N, Thatte U, Patankar S, Srivastava S. Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 7(8): e41751, 2012a.
- Ray S, Kamath KS, Srivastava R, Raghu D, Gollapalli K, Jain R, Gupta SV, Ray S, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Patankar S, Srivastava S. Serum proteome analysis of vivax malaria: An insight into the disease pathogenesis and host immune response. J Proteomics 75(10): 3063-80, 2012b.
- Srivastava R, Ray S, Vaibhav V, Gollapalli K, Jhaveri T, Taur S, Dhali S, Gogtay N, Thatte U, Srikanth R, Srivastava S. Serum profiling of leptospirosis patients to investigate proteomic alterations. J Proteomics 76: 56-68, 2012.
- Ray S, Srivastava R, Tripathi K, Vaibhav V, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS 16(10): 527-36, 2012c.
* Correspondence: Dr. Sanjeeva Srivastava, Department of Biosciences and Bioengineering, IIT Bombay, Mumbai 400 076, India: E-mail: sanjeeva@iitb.ac.in; Phone: +91-22-2576-7779, Fax: +91-22-2572-3480
![Figure 2 (a) Western blot analysis of haptoglobin (HP), serum amyloid A (SAA), and clusterin (CLU) from serum samples of healthy control (HC) [n = 12], falciparum malaria (FM) [n = 12], vivax malaria (VM) [n = 12], Leptospirosis (Lep) [n = 6], dengue fever [DF] [n = 6] and non infectious disease control (NIDC:GBM) [n = 12]. Representative blots of the target proteins are depicted along with their respective relative abundance volumes (volume X 104). All the data are represented as mean ± SE. (b) Discrimination of malaria from dengue, leptospirosis and GBM using PLS-DA analysis. PLS-DA scores Plot for FM (blue spheres, n = 8), VM (green spheres, n = 8), DF (red spheres, n = 6), Lep (grey spheres, n = 6) and GBM (brown spheres, n = 8) samples based on 6 differentially expressed proteins (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol-binding protein and apolipoprotein A-I) identified using DIGE. The axes of the plot indicate PLSDA latent variables t0-t2.](http://www.amritabioquest.org/conference/2013/wp-content/uploads/sites/2/2015/02/sanjeeva-2.jpg)



Abhijeet Kate, Arpana G Panicker, Diana Writer, Giridharan P, Keshav K V Ramamoorthy, Saji George, Shailendra K Sonawane
Protoplast fusion and transformation: A tool for activation of latent gene clusters
In the quest to discover new bioactive leads for unmet medical needs, actinomycetes present a treasure trove of undiscovered molecules. The ability of actinomycetes to produce antibiotics and other bioactive secondary metabolites has been underestimated due to sparse studies of cryptic gene clusters. These gene clusters can be tapped to explore scaffolds hidden in them. The up-regulation of the dormant genes is one of the most important areas of interest in the bioactive compounds discovery from microbial resources. Genome shuffling is a powerful tool for the activation of such gene clusters. Lei Yu, et al.1, reported enhancement of the lactic acid production in Lactobacillus rhamnosus through genome shuffling brought about by protoplast fusion. D. A. Hopwood et al.2 suggested that an interspecific recombination between strains producing different secondary metabolites, generate producers of ‘hybrid’ antibiotics. They also mentioned that an intraspecific fusion of actinomycetes protoplast bring about random and high frequency recombination. Protoplasts can also be used as recipients for isolated DNA, again in the presence of polyethylene glycol (PEG). In our study we had undertaken random genome shuffling by protoplast fusion of two, rather poorly expressed actinomycetes strains A (Figure 1) & B (Figure 2), mediated by PEG; and also by naked DNA transformation of Strain A protoplast with the DNA of Strain B. We generated eight protoplast fusants and seven transformants from parents considering their morphological difference from the two parent strains. These 15 recombinants were checked for their same colony morphologies for five generations to ensure phenotypic stability. Antibiotic resistance pattern was established by using antibiotic octodisc to generate a marker profile of the recombinants and the parent strains. Eight fusants (AP-18, AP-25, AP-2, AP-11, AP-14, AP-19, AP-11 and AP-27) and four transformants (TAP-30, TAP-31, TAP-32 and TAP-33) (Table 1) have shown a different antibiotic sensitivity pattern as compared to the parent strains. We envisage that these recombinants harbor shuffled gene clusters. To support array of conditions to express such shuffled/cryptic genes the recombinants were fermented in 11 different nutrient stress variants. The extracts generated were subjected to metabolite profiling by HPLC-ELSD, bioactivity screening for cytotoxicity and anti-infective capabilities. Two fusants AP-11 (Figure 3) and AP-25; one transformant TAP-32 (in growth media MBA-5 and MBA-7) displayed antifungal activity unlike parent strains (Table 2) Fusant AP-11 (Table 5) exhibited significant cell growth inhibition of five different cancer cell lines. The parents Strain A and Strain B did not exhibit any cell growth inhibition of these cell lines (Table 5). The metabolite profiling of fusant AP-11 and transformant TAP-32 was done by HPLC-ELSD. AP-11 showed the presence of five additional peaks (Figure 5 & Figure 6); TAP-32 extract from medium MBA-5 (Figure 7 & Figure 8) showed the presence of four additional peaks and TAP-32 extract from MBA-7 (Figure 9 & Figure 10) showed 14 additional peaks as compared to parent strains in similar medium and media controls. The study indicated that protoplast fusion and transformation have not only caused morphological changes but also shuffled genes responsible for synthesis of bioactive molecules. Further characterization of these new peaks is warranted.

Binu K Aa, Jem Prabhakarb, Thara Sc and Lakshmi Sd,∗
aDepartment of Clinical Diagnostics Services and Translational Research, Malabar Cancer Centre, Thalassery, Kerala, India.
bDivision of Surgical Oncology, Division of Pathology
dDivision of Cancer Research, Regional Cancer Centre, Thiruvananthapuram, Kerala, India.
Introduction
AIB1, a member of the nuclear co activators, promotes the transcriptional activity of multiple nuclear receptors such as the ER and other transcription factors. Chemokines produced by stromal cells have potential to influence ERα-positive breast cancer progression to metastasis. CXCR4 is the physiological receptor for SDF1, together shown to stimulate the chemotactic and invasive behavior of breast cancer cells to serve as a homing mechanism to sites of metastasis. We propose that over expression of AIB1 in breast cancer cells leads to increased SDF1 and CXCR4 expression, which induces invasion and metastasis of cancer cells.
Materials and Methods
Breast tumor and normal breast tissues from patients in Regional Cancer Centre, Thiruvananthapuram were used for study. The modulatory effect of AIB1 was studied in MCF-7 cells with AIB1 siRNA transfection along with treatment of 17β-Estradiol (E2), 4-hydroxytamoxifen (4OHT), combinations of E2 and 4OHT. The gene expression pattern and protein localization were assessed by RT-PCR and immunofluorescence microscopy respectively. The metastatic and invasive properties were assessed by wound healing assay. Quantitative colocalization analyses were done to assess the association of proteins using Pearson’s correlation coefficient.
Result and Conclusion
The mRNA and protein level expression of AIB1, CXCR4 and SDF1 were higher in tumor samples than in normal samples. AIB1 was localized to the nuclei whereas CXCR4 and SDF1 immunoreactivity were observed in the cytoplasm and to a lesser extent in the nuclei of tumor epithelial cells. In tumor samples the gene level expressions of AIB1 showed significant positive correlations with SDF1(r = 0.213, p = 0.018). CXCR4 showed significant positive correlation with SDF1 in gene (r = 0.498, p = 0.000) and protein levels(r = 0.375, p = 0.002). Quantitative colocalization analyses showed a marked reduction in expression of CXCR4 and SDF1 in siAIB1MCF-7 cells than MCF-7 cells with different treatment groups. Wound healing assay shows reduced wound healing in siAIB1 treated MCF-7 cells.
In recent years, targeting specific cancer pathways and key molecules to arrest tumor growth and achieve tumor eradication have proven a challenge; due to acquired resistance and homing of cancer cells to various metastatic sites. The present study revealed that silencing AIB1 can prevent the over expression of SDF1 and CXCR4. Co activator levels determine the basal and estrogen-inducible expression of SDF1, a secreted protein that controls breast cancer cell proliferation and invasion through autocrine and paracrine mechanisms (Hall et al. 2003). The effects of CXCR4 overexpression has been correlated with SDF1 mediated activation of downstream signaling via ERK1/2 and p38 MAPK and with an enhancement of ER-mediated gene expression (Rhodes et al. 2011). It is possible that over expression of AIB1 as a stimulant involved in the expression of CXCR4 might up-regulate the expression of prometastatic and angiogenic genes. Thus based on these observations it can be concluded that SDF1/CXCR4 overexpression, with significant association with AIB1 expression, itself contribute to the development of mammary cancer and metastatic progression.
 Nader Pourmand, Ph.D.
Nader Pourmand, Ph.D.
Director, UCSC Genome Technology Center,University of California, Santa Cruz
Biosensor and Single Cell Manipulation using Nanopipettes
Approaching sub-cellular biological problems from an engineering perspective begs for the incorporation of electronic readouts. With their high sensitivity and low invasiveness, nanotechnology-based tools hold great promise for biochemical sensing and single-cell manipulation. During my talk I will discuss the incorporation of electrical measurements into nanopipette technology and present results showing the rapid and reversible response of these subcellular sensors to different analytes such as antigens, ions and carbohydrates. In addition, I will present the development of a single-cell manipulation platform that uses a nanopipette in a scanning ion-conductive microscopy technique. We use this newly developed technology to position the nanopipette with nanoscale precision, and to inject and/or aspirate a minute amount of material to and from individual cells or organelle without comprising cell viability. Furthermore, if time permits, I will show our strategy for a new, single-cell DNA/ RNA sequencing technology that will potentially use nanopipette technology to analyze the minute amount of aspirated cellular material.
 Michelle Hermiston, MD, Ph.D.
Michelle Hermiston, MD, Ph.D.
Assistant Professor, Department of Pediatrics University of California San Francisco, USA
Interrogating Signaling Networks at the Single Cell Level In Primary Human Patient Samples
Multiparameter phosphoflow cytometry is a highly sensitive proteomic approach that enables monitoring of biochemical perturbations at the single cell level. By combining antisera to cell surface markers and key intracellular proteins, perturbations in signaling networks, cell survival and apoptosis mediators, cell cycle regulators, and/or modulators of other cellular processes can be analyzed in a highly reproducible and sensitive manner in the basal state and in response to stimulation or drug treatment. Advantages of this approach include the ability to identify the biochemical consequences of genetic and/or epigenetic changes in small numbers of cells, to map potential interplay between various signaling networks simultaneously in a single cell, and to interrogate potential mechanisms of drug resistance or response in a primary patient sample. Application of this technology to patients with acute lymphoblastic leukemia or the autoimmune disease systemic lupus erythematosus (SLE) will be discussed.
 Lalitha Subramanian, Ph.D.
Lalitha Subramanian, Ph.D.
Chief Scientific Officer & VP, Services at Scienomics, USA
Nanoscale Simulations – Tackling Form and Formulation Challenges in Drug Development and Drug Delivery
Lalitha Subramanian, Dora Spyriouni, Andreas Bick, Sabine Schweizer, and Xenophon Krokidis Scienomics
The discovery of a compound which is potent in activity against a target is a major milestone in Pharmaceutical and Biotech industry. However, a potent compound is only effective as a therapeutic agent when it can be administered such that the optimal quantity is transported to the site of action at an optimal rate. The active pharmaceutical ingredient (API) has to be tested for its physicochemical properties before the appropriate dosage form and formulation can be designed. Some of the commonly evaluated parameters are crystal forms and polymorphs, solubility, dissolution behavior, stability, partition coefficient, water sorption behavior, surface properties, particle size and shape, etc. Pharmaceutical development teams face the challenge of quickly and efficiently determining a number of properties with small quantities of the expensive candidate compounds. Recently the trend has been to screen these properties as early as possible and often the candidate compounds are not available in sufficient quantities. Increasingly, these teams are leveraging nanoscale simulations similar to those employed by drug discovery teams for several decades. Nanoscale simulations are used to predict the behavior using very little experimental data and only if this is promising further experiments are done. Another aspect where nanoscale simulations are being used in drug development and drug delivery is to get insights into the behavior of the system so that process failures can be remediated and formulation performance can be improved. Thus, the predictive screening and the in-depth understanding leads to experimental efficiency resulting in far-reaching business impacts.
With specific examples, this talk will focus on the different types of nanoscale simulations used to predict properties of the API in excipients and also provide insight into system behavior as a function of shelf life, temperature, mechanical stress, etc.
 Srisairam Achuthan, Ph.D.
Srisairam Achuthan, Ph.D.
Senior Scientific Programmer, Research Informatics Division, Department of Information Sciences, City of Hope, CA, USA
Applying Machine learning for Automated Identification of Patient Cohorts
Srisairam Achuthan, Mike Chang, Ajay Shah, Joyce Niland
Patient cohorts for a clinical study are typically identified based on specific selection criteria. In most cases considerable time and effort are spent in finding the most relevant criteria that could potentially lead to a successful study. For complex diseases, this process can be more difficult and error prone since relevant features may not be easily identifiable. Additionally, the information captured in clinical notes is in non-coded text format. Our goal is to discover patterns within the coded and non-coded fields and thereby reveal complex relationships between clinical characteristics across different patients that would be difficult to accomplish manually. Towards this, we have applied machine learning techniques such as artificial neural networks and decision trees to determine patients sharing similar characteristics from available medical records. For this proof of concept study, we used coded and non-coded (i.e., clinical notes) patient data from a clinical database. Coded clinical information such as diagnoses, labs, medications and demographics recorded within the database were pooled together with non-coded information from clinical notes including, smoking status, life style (active / inactive) status derived from clinical notes. The non-coded textual information was identified and interpreted using a Natural Language Processing (NLP) tool I2E from Linguamatics.

Tejaswini Subbannayya, Nandini A. Sahasrabuddhe, Arivusudar Marimuthu, Santosh Renuse, Gajanan Sathe, Srinivas M. Srikanth, Mustafa A. Barbhuiya, Bipin Nair, Juan Carlos Roa, Rafael Guerrero-Preston, H. C. Harsha, David Sidransky, Akhilesh Pandey, T. S. Keshava Prasad and Aditi Chatterjee
Proteomic profiling of gallbladder cancer secretome – a source for circulatory biomarker discovery
Gallbladder cancer (GBC) is the fifth most common cancer of the gastrointestinal tract and one of the common malignancies that occur in the biliary tract (Misra et al. 2006; Lazcano-Ponce et al. 2001). It has a poor prognosis with survival of less than 5 years in 90% of the cases (Misra et al. 2003). The etiology is ill-defined. Several risk factors have been reported including cholelithiasis, obesity, female gender and exposure to carcinogens (Eslick 2010; Kumar et al. 2006). Poor prognosis in GBC is mainly due to late presentation of the disease and lack of reliable biomarkers for early diagnosis. This emphasizes the need to identify and characterize cancer biomarkers to aid in the diagnosis and prognosis of GBC. Secreted proteins are an important class of molecules which can be detected in body fluids and has been targeted for biomarker discovery. There are challenges faced in the proteomic interrogation of body fluids especially plasma such as low abundance of tumor secreted proteins, high complexity and high abundance of other proteins that are not released by the tumor cells (Tonack et al. 2009). Profiling of conditioned media from the cancer cell lines can be used as an alternate means to identify secreted proteins from tumor cells (Kashyap et al. 2010; Marimuthu et al. 2012). We analyzed the invasive property of 7 GBC cell lines (SNU-308, G-415, GB-d1, TGBC2TKB, TGBC24TKB, OCUG-1 and NOZ). Four cell lines were selected for analysis of the cancer secretome based on the invasive property of the cells. We employed isobaric tags for relative and absolute quantitation (iTRAQ) labeling technology coupled with high resolution mass spectrometry to identify and characterize secretome from the panel of 4GBC cancer cells mentioned above. In total, we have identified around 2,000 proteins of which 175 were secreted at differential abundance across all the four cell lines. This secretome analysis will act as a reservoir of candidate biomarkers. Currently, we are investigating and validating these candidate markers from GBC cell secretome. Through this study, we have shown mass spectrometry-based quantitative proteomic analysis as a robust approach to investigate secreted proteins in cancer cells.